S-1/A: General form of registration statement for all companies including face-amount certificate companies
Published on December 4, 2023
As filed with the Securities and Exchange Commission on December 1, 2023.
Registration No. 333-275361
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 3 to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
FibroBiologics, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 86-3329066 | ||
|
(State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
455 E. Medical Center Blvd.
Suite 300
Houston, Texas 77598
(281) 671-5150
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Pete O’Heeron
Chief Executive Officer
FibroBiologics, Inc.
455 E. Medical Center Blvd.
Suite 300
Houston, Texas 77598
(281) 671-5150
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
|
Brian Fenske Norton Rose Fulbright US LLP 1301 McKinney Street Suite 5100 Houston, Texas 77010 (713) 651-5151 |
|
Pete O’Heeron Chief Executive Officer FibroBiologics, Inc. 455 E. Medical Center Blvd. Suite 300 Houston, Texas 77598 (281)
671-5150 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after this registration statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box. ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ |
| Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion, dated , 2023
Shares

FibroBiologics, Inc.
Common Stock
This prospectus relates to the registration of the resale of up to shares of our common stock by our stockholders identified in this prospectus, or the Registered Stockholders, in connection with our direct listing, or the Direct Listing, on the Nasdaq Global Market, or Nasdaq. Unlike an initial public offering, the resale by the Registered Stockholders is not being underwritten on a firm-commitment basis by any investment bank. The Registered Stockholders may, or may not, elect to sell their shares of common stock covered by this prospectus, as and to the extent they may determine. The Registered Stockholders may offer, sell or distribute all or a portion of the shares of common stock hereby registered publicly or through private transactions at prevailing market prices or at negotiated prices. If the Registered Stockholders choose to sell their shares of common stock, we will not receive any proceeds from the sale of shares of common stock by the Registered Stockholders.
Our board of directors and our stockholders have each approved on October 6, 2023 a 1-for-4 reverse stock split of all classes of our issued and outstanding capital stock, or the Reverse Stock Split. On October 31, 2023, we filed an amended and restated certificate of incorporation with the State of Delaware to immediately effect the Reverse Stock Split. All share and per share information in this prospectus have been adjusted to reflect the Reverse Stock Split, unless otherwise stated.
No public market for our common stock currently exists, and our shares of common stock have a limited history of trading in private transactions. In December 2022, we issued an aggregate of the equivalent of 381,658 shares of Series B Preferred Stock to investors in a private placement, at a price of the equivalent of $6.76 per share as to the equivalent of 318,049 shares, with the remaining equivalent of 63,609 shares being bonus shares. From February 2023 through April 2023, we issued an aggregate of the equivalent of 890,310 shares of Series B Preferred Stock to investors in a Regulation Crowdfunding offering, at a price of the equivalent of $6.76 per share as to the equivalent of 724,937 shares, with the remaining equivalent of 143,225 shares and equivalent of 22,148 shares being bonus shares and commission payment shares, respectively. In March and April 2023, we issued the equivalent of 1,680,084 shares of Series B Preferred Stock to investors in private placements, at a price of the equivalent of $6.76 per share as to the equivalent of 1,527,349 shares, with the remaining equivalent of 152,735 shares being bonus shares. In April 2023 through September 2023, we issued the equivalent of 74,922 shares of Series B-1 Preferred Stock to investors in a private placement, at prices ranging from the equivalent of $18.00 to the equivalent of $20.00 per share as to the equivalent of 64,070 shares, with the remaining equivalent of 10,852 shares being bonus shares. In connection with a portion of such private placement of our Series B-1 Preferred Stock, we also agreed to issue warrants, exercisable for a period of three years from our Direct Listing, to purchase an aggregate of the equivalent of an aggregate of 8,890 shares of our common stock at an exercise price of the equivalent of $20.00 per share. In November 2023, the Company issued a total of 14,859 additional shares of Series B-1 Preferred Stock and 1,431 additional warrants to purchase shares of common stock to investors who subscribed to purchase shares of Series B-1 Preferred Stock at a price per share that exceeded the reference price per share expected in the Direct Listing.
Upon the Direct Listing, all of our then outstanding shares of our Series B Preferred Stock and Series B-1 Preferred Stock will automatically convert, without the payment of additional consideration by or to the holders thereof, into shares of our common stock on a one-for-one basis.
Recent purchase prices of our common stock in private transactions may have little or no relation to the opening public price of our shares of common stock on Nasdaq or the subsequent trading price of our shares of common stock on Nasdaq. For more information, see “Sale Price History of Our Capital Stock.” Further, the listing of our common stock on Nasdaq, without a firm-commitment underwritten offering, is a novel method for commencing public trading in shares of our common stock and, consequently, the trading volume and price of shares of our common stock may be more volatile than if shares of our common stock were initially listed in connection with an initial public offering underwritten on a firm-commitment basis.
On the day that our shares of common stock are initially listed on Nasdaq, Nasdaq will begin accepting, but not executing, pre-opening buy and sell orders and will begin to continuously generate the indicative Current Reference Price (as defined below) on the basis of such accepted orders. The Current Reference Price is calculated each second and, during a 10-minute “Display Only” period, is disseminated, along with other indicative imbalance information, to market participants by Nasdaq on its NOII and BookViewer tools. Following the “Display Only” period, a “Pre-Launch” period begins, during which Maxim Group LLC, or the Advisor, in its capacity as our financial advisor, must notify Nasdaq that our shares are “ready to trade.” Once the Advisor has notified Nasdaq that our shares of common stock are ready to trade, Nasdaq will confirm the Current Reference Price for our shares of common stock, in accordance with Nasdaq rules. If the Advisor then approves proceeding at the Current Reference Price, the applicable orders that have been entered will be executed at such price and regular trading of our shares of common stock on Nasdaq will commence, subject to Nasdaq conducting validation checks in accordance with Nasdaq rules. Under Nasdaq rules, the “Current Reference Price” means: (i) the single price at which the maximum number of orders to buy or sell can be matched; (ii) if there is more than one price at which the maximum number of orders to buy or sell can be matched, then it is the price that minimizes the imbalance between orders to buy or sell (i.e. minimizes the number of shares that would remain unmatched at such price); (iii) if more than one price exists under (ii), then it is the entered price (i.e. the specified price entered in an order by a customer to buy or sell) at which our shares of common stock will remain unmatched (i.e. will not be bought or sold); and (iv) if more than one price exists under (iii), a price determined by Nasdaq in consultation with the Advisor in its capacity as our financial advisor. In the event that more than one price exists under (iii), the Advisor will exercise any consultation rights only to the extent that it can do so consistent with the anti-manipulation provisions of the federal securities laws, including Regulation M, or applicable relief granted thereunder. The Registered Stockholders will not be involved in Nasdaq’s price-setting mechanism, including any decision to delay or proceed with trading, nor will they control or influence the Advisor in carrying out its role as a financial adviser. The Advisor will determine when our shares of common stock are ready to trade and approve proceeding at the Current Reference Price primarily based on considerations of volume, timing and price. In particular, the Advisor will determine, based primarily on pre-opening buy and sell orders, when a reasonable amount of volume will cross on the opening trade such that sufficient price discovery has been made to open trading at the Current Reference Price. For more information, see “Plan of Distribution” beginning on page 126 of this prospectus.
We have applied to list our common stock on the Nasdaq Global Market under the symbol “FBLG.” We expect our common stock to begin trading on Nasdaq on or about , 2023.
If our Nasdaq application is not approved or we otherwise determine that we will not be able to secure the listing of our common stock on Nasdaq, we will not complete this Direct Listing. This listing is a condition to the offering. No assurance can be given that our Nasdaq application will be approved and that our common stock will ever be listed on Nasdaq. If our listing application is not approved by Nasdaq, we will not be able to consummate the offering and we will terminate this Direct Listing.
Upon completion of this offering, our founder and Chief Executive Officer, Pete O’Heeron, will collectively beneficially own approximately 59% of the voting power of our outstanding voting securities and we will be a “controlled company” within the meaning of the listing rules of The Nasdaq Stock Market LLC. We do not intend to rely on any exemptions from the corporate governance requirements that are available to controlled companies.
We are an “emerging growth company” and a “smaller reporting company” as defined under the federal securities laws and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus and may elect to do so in future filings. See “Prospectus Summary—Implications of Being an Emerging Growth Company and a Smaller Reporting Company.”
Investing in our common stock involves a high degree of risk. See the “Risk Factors” section beginning on page 9 of this prospectus for the risks and uncertainties you should consider before investing in our common stock.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Prospectus dated , 2023
TABLE OF CONTENTS
You should rely only on the information contained in this prospectus or contained in any free writing prospectus filed with the Securities and Exchange Commission. Neither we nor any of the Registered Stockholders have authorized anyone to provide any information different from, or in addition to, the information contained in this prospectus and in any free writing prospectuses we have prepared or that have been prepared on our behalf or to which we have referred you. Neither we nor any of the Registered Stockholders take responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. The Registered Stockholders are offering to sell, and seeking offers to buy, shares of their common stock only under the circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus is current only as of its date, regardless of the time of delivery of this prospectus or of any sale of our common stock. Our business, financial condition, results of operations and prospects may have changed since such date.
For investors outside the United States: Neither we nor any of the Registered Stockholders have done anything that would permit the use of or possession or distribution of this prospectus or any related free writing prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the shares of our common stock by the Registered Stockholders and the distribution of this prospectus outside the United States.
| i |
This prospectus is a part of a registration statement on Form S-1 that we filed with the Securities and Exchange Commission, or the SEC, using a “shelf” registration or continuous offering process. Under this process, the Registered Stockholders may, from time to time, sell the common stock covered by this prospectus in the manner described in the section titled “Plan of Distribution.” Additionally, we may provide a prospectus supplement to add information to, or update or change information contained in, this prospectus, including the section titled “Plan of Distribution”. You may obtain this information without charge by following the instructions under the “Where You Can Find Additional Information” section of this prospectus. You should read this prospectus and any prospectus supplement before deciding to invest in our common stock.
This prospectus contains summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information. All of the summaries are qualified in their entirety by the actual documents. Copies of some of the documents referred to herein have been filed or will be filed as exhibits to the registration statement of which this prospectus is a part, and you may obtain copies of those documents as described under “Where You Can Find Additional Information.”
| 1 |
This summary highlights select information contained elsewhere in this prospectus and does not contain all the information you should consider before making an investment decision. You should read the entire prospectus carefully, including the sections entitled “Risk Factors,” “Cautionary Note Regarding Forward-Looking Statements,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and the accompanying notes included elsewhere in this prospectus before making an investment decision. Unless otherwise indicated or the context otherwise requires, all references in this prospectus to “we,” “us,” “our,” the “Company,” “FibroBiologics” and similar terms refer to FibroBiologics, Inc.
Overview
We are a clinical-stage cell therapy company focused on developing and commercializing fibroblast-based therapies for patients suffering from chronic diseases with significant unmet medical needs, including degenerative disc disease, multiple sclerosis, wound healing, and certain cancers, and for potential extension of life applications including thymic and splenic involution reversal.
We were formed in April 2021 as a Texas limited liability company under the name FibroBiologics, LLC, and converted to a Delaware corporation in December 2021 under the name Fibrobiologics, Inc. On April 12, 2023, we changed our name to FibroBiologics, Inc. In connection with our formation, we issued shares of our Series A Preferred Stock, or the Series A Preferred Stock, to our then parent, SpinalCyte LLC (doing business as FibroGenesis), or FibroGenesis, in return for rights to certain intellectual property through a patent assignment agreement and an intellectual property cross-licensing agreement. Developing the intellectual property obtained from FibroGenesis was the basis for our formation. Prior to our inception, preclinical research and development related to the transferred intellectual property took place under FibroGenesis.
Fibroblasts
Technology Platform
Fibroblasts and stem cells are the only two cell types in the human body that can regenerate tissue and organs. Studies have indicated that mesenchymal stem cells and fibroblasts share many surface markers in common, and can differentiate into many cells including adipocytes, chondrocytes, osteoblasts, hepatocytes, and cardiomyocytes, and can regulate the immune system. However, transcriptomic and epigenetic studies have indicated a clear difference between the two cell types.
Fibroblasts comprise the main cell type of connective tissue, possessing a spindle-shaped morphology, whose classical function has historically been believed to produce an extracellular matrix responsible for maintaining the structural integrity of the tissue. Fibroblasts also play an important role in maintaining stem cell niches in organs and are involved in every stage of wound healing.
Fibroblasts are favorable to stem cells as a cell therapy treatment platform because fibroblasts:
| ● | can be non-invasively harvested from a variety of skin donors from surgical procedures such as tummy tuck flaps or simple biopsy punch; |
| ● | have a faster doubling time in culture than stem cells; |
| ● | possess superior immune modulatory activity compared with stem cells; |
| ● | exhibit enhanced ability to produce regenerative cytokines and growth factors compared with stem cells; and |
| ● | are more economical to isolate, culture and expand compared with stem cells because fibroblasts do not require the use of expensive tissue culture media and additives. |
Studies have demonstrated that allogeneic fibroblasts, much like mesenchymal stem cells, are immune-privileged and do not provoke an immune response in vitro and in vivo. If autologous fibroblasts were required instead, it would mean that cells would have to be harvested from each patient, processed and cultured, and then administered to the same patient, which would be more costly and inefficient. Because allogeneic fibroblasts do not cause an immune response, we are planning to build our own current Good Manufacturing Practices, or cGMP, manufacturing facility to source allogeneic fibroblast cells for clinical testing of our product candidates and for commercial sales if our product candidates receive marketing approval.
To date, however, no fibroblast therapy products have been approved and there have only been a few clinical trials involving fibroblasts. The costs to develop, manufacture, and commercialize product candidates utilizing our fibroblasts technology platform may exceed our estimates. Furthermore, the biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary and novel products and product candidates so any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. Additional information regarding risks and uncertainties relating to our product candidates technology and business are set forth in the sections titled “—Summary of Risk Factors” and “Risk Factors” in this prospectus.
| 2 |
Our Management Team and Oversight
We have assembled an executive leadership team comprised of our founder, chief executive officer and chairperson of our board of directors, our chief scientific officer, and our chief financial officer, with successful track records in startup entrepreneurial companies and in the life sciences industry. Our executive leadership team works under the oversight of our board of directors who are recognized leaders with hands-on industry experience. We also have a team of world-renowned scientists with relevant expertise on our scientific advisory board to help guide our research and development efforts.
Our Current Pipeline
We have a pipeline of product candidates at various stages of development, including the following:
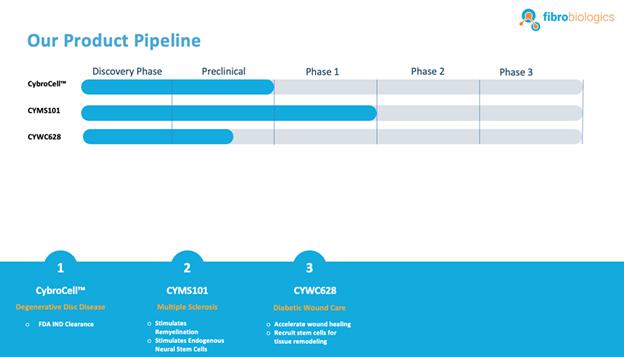
CybroCell™ for Degenerative Disc Disease: CybroCell™ is an allogeneic fibroblast cell-based therapy for degenerative disc disease This new technology is being designed as an alternative method for repairing the cartilage of the intervertebral disc (or any other articular cartilage). The method is based on using human dermal fibroblasts, or HDFs, which are forced to differentiate into chondrocyte-like cells in vivo using the mechanical force and intermittent hydrostatic pressure found in the spine, for chondrogenic differentiation of fibroblasts. We believe our solution will prove superior to existing treatments because we expect it will be less invasive, and will regenerate the disc, restore function and reduce pain without debilitating long-term effects. We have completed two rounds of animal studies. The results from the studies were positive and resulted in “first in human” trial approval in our investigational new drug, or IND, submission to the U.S. Food and Drug Administration, or FDA. We have received IND clearance from the FDA, conditional upon approval of our master cell bank, to run a Phase 1/2 clinical trial for patients suffering from degenerative disc disease. We will be conducting this trial within the United States. A timeline will be determined through discussions with the FDA.
| 3 |
CYMS101 for Multiple Sclerosis: We are developing CYMS101 as an allogeneic fibroblast cell-based therapy to treat multiple sclerosis, or MS. After completing animal studies using CYMS101 (allogeneic fibroblast cells), we received approval from Mexico to conduct clinical investigations using the fibroblast cell composition for patients with MS and have completed a Phase 1 clinical trial called “Feasibility Study of Tolerogenic Fibroblasts in Patients with Refractory Multiple Sclerosis.” The study was conducted in five participants. The primary objective of the study was to assess safety, and the secondary objective was to assess efficacy. The results of the study for safety were no adverse effects during intravenous injection of the tolerogenic fibroblasts, no short or long-impact in complete blood count test during the 16-week monitoring period, and no short or long impact in electrocardiogram results during the 16-week monitoring period. In addition, the results of the study for efficacy included general improvement of Paced Auditory Serial Addition Test, or PASAT, score for all patients during the 16-week monitoring period, general improvement of 9-hole Peg test completion time for all patients during the 16-week testing period, no general improvement or deterioration noted with the Timed 25-Foot walk test, no general improvement or deterioration noted with Expanded Disability Status Scale, or EDSS, test, and no patient exhibited further deterioration during the trial. We are currently conducting further research to determine the mode of action of fibroblasts in oligodendrocyte expansion and expect to file an IND application for a Phase 2 clinical trial in MS. We will likely seek a strategic partner to collaborate with us on the development of CYMS101 either before initiating the Phase 2 clinical trial, or after its completion, if successful, and prior to commencing with a Phase 3 clinical trial.
CYWC628 for Wound Healing: We are in the late pre-clinical stages of developing CYWC628 as an allogeneic fibroblast cell-based therapy for wound healing. Our studies are presently focused on utilizing fibroblasts and fibroblast-derived cells to treat wounds in diabetic mice. Our data to date is compiled from four separate animal model studies (manuscript for publication in progress). Each study utilized 16 wild type as well as leptin mutated NONcNZO10LTJ mouse that develops type 2 diabetes when fed a high fat diet. Wound size and area for all our experiments were measured using an eKare inSight™ device which is FDA approved for measuring and monitoring wound size, area and depth. Phase 1 of our pre-clinical study studied the subcutaneous and topically administered single cell mouse dermal fibroblasts (both treatments administered every two days), as well as mouse dermal fibroblast derived exosomes. The results of this study indicated significant improvement in wound healing (p <0.0005) for topically administered mouse fibroblasts and mouse fibroblast exosomes as compared to untreated control, and significant improvement in wound healing with subcutaneous inject of fibroblast in the wound periphery (p < .005). Our phase 2 pre-clinical study studied the impact of using frozen and thawed single cell mouse fibroblasts administered every two days, as well as mouse spheroid fibroblasts, one-time topical administration, measuring 250 um and each containing approximately 10,000 mouse dermal fibroblasts. In total 100 spheroids were topically administered on to an 8 millimeter diameter wound on the back of the wild type and leptin mutated mice. The results of the study indicated significant improvement in wound healing with the frozen thawed single cell mouse fibroblasts (p < 0.005), as well as 4°C stored mouse fibroblast spheroids (p <0.0005) with both mouse types. Our objective was to test the feasibility of using spheroid fibroblasts as an extended-release mechanism on wound surfaces. The results indicated that spheroid fibroblasts are easier, do not require cold chain logistics, and are more viable to use, in addition to generating more significant results. Our phase 3 pre-clinical study tested the effect of using a single topical administration of human dermal fibroblast (CYWC628) spheroids compared to a single administration of mouse dermal spheroids, in addition to comparing with a commercially available and FDA approved diabetic foot ulcer treatment called Grafix™. The results of our study indicated that CYWC628 significantly improved wound healing rate (p < 0.0005) as compared to untreated control as well as significant improvement (p < 0.05) over mouse fibroblast spheroids and Grafix™. For our Phase 4 pre-clinical study we studied the impact of a single topical treatment of CYWC628 spheroids and Grafix™ on a chemically induced chronic wound model often used to mimic diabetic foot ulcers in animal models. The results of our study indicated a 58.5% reduction in wound area three days after a single topical administration of CYWC628 as compared to 34.5% for Grafix™ (p < 0.005). The untreated saline control group had an 11% improvement in wound healing which was not statistically significant (p < 0.06). Our results also indicated that with multiple topical administration of CYWC628, the rate of wound closure will likely be more rapid. For our last pre-clinical study, we will investigate multiple administrations of CYWC628 on a chemically induced chronic wound mouse model to provide information on frequency of CYWC628 administration. We expect to complete this study in the fourth quarter of 2023. Based upon our results achieved to date, we plan to pursue an IND submission with the FDA for wound healing as early as 2024.
Our Competitive Strengths
Our strengths lie in our technology platform centered around the power of fibroblasts and in our experienced leadership team. Fibroblasts are the most common cell found in the human body and we believe they are more robust and potent than stem cells. Our intellectual property portfolio includes 48 issued patents and 108 pending patents for the use of fibroblasts in diverse therapeutic areas. We also have an experienced leadership team with successful track records in entrepreneurial startup companies and the life sciences industry, a board of directors with life sciences operational leadership experience, and a world-renowned scientific advisory board with relevant expertise.
Our Strategy
We are leveraging fibroblast cells as a technology platform to research and develop innovative treatments for chronic diseases with significant unmet treatment needs. Our vision is to become a world leader in regenerative medicine through a rigorous scientific process and commitment to serving patients’ needs. To achieve our vision, we will focus our efforts on the following strategy:
| ● | Prioritize our initial clinical development efforts on product candidates with the combination of significant unmet treatment needs, lower risk and high market potential. |
| 4 |
| ● | Partner with contract research organizations, or CROs, with the relevant expertise and experience to successfully and timely execute clinical trials to generate reliable pivotal data that can be used to seek approvals. | |
| ● | Attract and retain scientists with the skill sets required to conduct preclinical studies and identify the optimal paths forward to clinical trials. |
| ● | Invest in critical capabilities required to produce and supply fibroblasts for clinical trials and initial commercialization. |
| ● | Protect, expand and defend our intellectual property portfolio around fibroblasts. |
| ● | Expand development efforts in product candidates with longer development timelines, greater risk and significant unmet treatment needs as funding allows. |
Summary of Risk Factors
Our business is subject to numerous risks and uncertainties that you should be aware of before making an investment decision, including those highlighted in the section entitled “Risk Factors” in this prospectus. These risks include, but are not limited to, the following:
| ● | The successful development of biopharmaceutical products is highly uncertain. |
| ● | We have a limited operating history and none of our current product candidates have been approved for commercial sale. |
| ● | We have incurred significant net losses since inception, expect to continue to incur significant net losses for the foreseeable future and may never achieve or maintain profitability. |
| ● | We will require substantial additional capital to finance our operations. If we are unable to raise such capital when needed, or on acceptable terms, we may be forced to delay, reduce and/or eliminate one or more of our research and drug development programs or future commercialization efforts. |
| ● | The regulatory approval processes of the FDA, the European Medicines Agency, or the EMA, and other comparable foreign regulatory authorities are lengthy, time consuming and inherently unpredictable. |
| ● | We may encounter substantial delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates. |
| ● | The outcome of preclinical studies or early clinical trials may not be predictive of the success of later clinical trials, and the results of our clinical trials may not satisfy the requirements of the FDA, the EMA or other comparable foreign regulatory authorities. |
| ● | Interim, topline and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data. |
| ● | Our current or future product candidates may cause adverse events, toxicities or other undesirable side effects when used alone or in combination with other approved products or investigational new drugs that may result in a safety profile that could inhibit regulatory approval, prevent market acceptance, limit their commercial potential or result in significant negative consequences. |
| ● | Even if approved, our product candidates may not achieve adequate market acceptance. |
| ● | Our refrigerated product candidates require specific storage, handling and administration at the clinical sites. |
| ● | We intend to identify and develop novel cell therapy product candidates, which makes it difficult to predict the time, cost and potential success of product candidate development. |
| 5 |
| ● | Because cell therapy is novel and the regulatory landscape that governs any cell therapy product candidates we may develop is rigorous, complex, uncertain and subject to change, we cannot predict the time and cost of obtaining regulatory approval, if we receive it at all, for any product candidates we may develop. |
| ● | We may be unable to obtain U.S. or foreign regulatory approvals and, as a result, may be unable to commercialize our product candidates. |
| ● | Any product candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated. |
| ● | We have limited experience in designing clinical trials. |
| ● | Our long-term prospects depend in part upon discovering, developing and commercializing additional product candidates, which may fail in development or suffer delays that adversely affect their commercial viability. |
| ● | We have never commercialized a fibroblast cell-based therapy product candidate before and may lack the necessary expertise, personnel and resources to successfully commercialize any product candidates on our own or together with suitable collaborators. |
| ● | We face significant competition. |
| ● | If we are unable to establish sales or marketing capabilities or enter into agreements with third parties to sell or market our product candidates, we may not be able to successfully sell or market our product candidates that obtain regulatory approval. |
| ● | In order to successfully implement our plans and strategies, we will need to grow the size of our organization, and we may experience difficulties in managing this growth. |
| ● | We are subject to risks related to our dependence on third parties (i) to conduct certain aspects of our preclinical studies and clinical trials and (ii) for certain portions of our manufacturing process. |
| ● | We are highly dependent on our Houston, Texas facility and any failure to maintain the use of this facility would have a material and adverse effect on our business. |
| ● | We are subject to extensive government regulations. |
| ● | Our business entails a significant risk of product liability. |
| ● | The FDA, the EMA and other comparable foreign regulatory authorities may not accept data from trials conducted in locations outside of their jurisdiction. |
| ● | Even if our product candidates receive regulatory approval, they will be subject to significant post-marketing regulatory requirements and oversight. |
| ● | Our success depends on our ability to protect our intellectual property and our proprietary technologies, and we are subject to various risks relating to our intellectual property. |
| ● | Our listing differs significantly from a firm-commitment underwritten initial public offering. | |
| ● | The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain executive management and qualified board members. | |
| ● | We will be a “controlled company” within the meaning of the Nasdaq Stock Market Rules upon the Direct Listing because our insiders will beneficially own more than 50% of the voting power of our outstanding voting securities. | |
| ● | Upon the Direct Listing, we will have 2,500 shares of Series C Preferred Stock with super voting rights. | |
| ● | We have identified a material weakness in our internal controls over financial reporting due to lack of segregation of duties. |
| ● | Our shares of common stock currently have no public market. An active trading market may not develop or continue to be liquid and the market price of our shares of common stock may be volatile. |
Reverse Stock Split
On October 6, 2023, our board of directors and our stockholders each approved the 1-for-4 Reverse Stock Split, and on October 31, 2023, we filed an amended and restated certificate of incorporation with the State of Delaware to immediately effect the Reverse Stock Split. All share and per share information in this prospectus have been adjusted to reflect the Reverse Stock Split, unless otherwise stated.
| 6 |
Adjustments to Authorized Capital Stock
In connection with the Reverse Stock Split, our board of directors and stockholders have also approved reductions in the number of capital stock, and the respective securities constituting our capital stock, we are authorized to issue.
Immediately prior to the Reverse Stock Split, the total number of shares of all classes of capital stock that we were authorized to issue was 600,000,000 shares, consisting of (i) 400,000,000 shares of voting common stock (which we sometimes refer to in this prospectus as our “common stock”), (ii) 120,000,000 shares of non-voting common stock and (iii) 80,000,000 shares of preferred stock, of which 35,000,000 were designated as Series A Preferred Stock, 20,000,000 were designated as Series B Preferred Stock, 20,000,000 were designated as Series B-1 Preferred Stock and 10,000 are designated as Series C Preferred Stock.
Pursuant to the adjustments to our authorized capital stock, immediately after the Reverse Stock Split, the total number of shares of all classes of capital stock that we are authorized to issue is 150,000,000 shares, consisting of (i) 100,000,000 shares of voting common stock (which we sometimes refer to in this prospectus as our “common stock”), (ii) 30,000,000 shares of non-voting common stock and (iii) 20,000,000 shares of preferred stock, of which 8,750,000 shares are designated as Series A Preferred Stock, 5,000,000 shares are designated as Series B Preferred Stock, 5,000,000 shares are designated as Series B-1 Preferred Stock and 2,500 shares are designated as Series C Preferred Stock. We sometimes refer to the foregoing adjustments in our capital stock in this prospectus as the “Authorized Capital Stock Adjustments.”
Upon consummation of the Direct Listing, after giving effect to the filing and effectiveness of our amended and restated certificate of incorporation and the adoption of our amended and restated bylaws, we will be authorized to issue 110,000,000 shares of capital stock, which will consist of: (i) 100,000,000 shares of common stock, par value $0.00001 per share and (ii) 10,000,000 shares of preferred stock, par value $0.00001 per share, of which 2,500 shares are designated as Series C Preferred Stock. See “Description of Capital Stock” for additional details.
Implications of being a Controlled Company
Upon completion of the Direct Listing, our founder and Chief Executive Officer, Pete O’Heeron, will collectively beneficially own approximately 59% of the voting power of our outstanding voting securities and we will be a “controlled company” within the meaning of the listing rules of The Nasdaq Stock Market LLC.
As long as our principal shareholder owns at least 50% of the voting power of our Company, we will be a “controlled company” as defined under Nasdaq Listing Rules. As a controlled company, we are permitted to rely on certain exemptions from Nasdaq’s corporate governance rules, including:
| ● | an exemption from the rule that a majority of our board of directors must be independent directors; |
| ● | an exemption from the rule that the compensation of our chief executive officer must be determined or recommended solely by independent directors; and |
| ● | an exemption from the rule that our director nominees must be selected or recommended solely by independent directors. |
Although we currently do not intend to rely on the “controlled company” exemption under the Nasdaq listing rules, we could elect to rely on this exemption in the future. As a result, you may not in the future have the same protection afforded to shareholders of companies that are subject to these corporate governance requirements.
Implications of being an emerging growth company and a smaller reporting company
We are an “emerging growth company” as defined in the Securities Act of 1933, or the Securities Act, as modified by the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. As such, we are eligible to take, and intend to take, advantage of certain exemptions from various reporting requirements applicable to other public companies that are not emerging growth companies for as long as we continue to be an emerging growth company, including (i) the exemption from the auditor attestation requirements with respect to internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, (ii) the exemptions from say-on-pay, say-on-frequency and say-on-golden parachute voting requirements and (iii) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements.
We will remain an emerging growth company until the earliest of (i) December 31, 2028, (ii) the last day of the fiscal year in which we have total annual gross revenue of at least $1.235 billion, (iii) the last day of the fiscal year in which we are deemed to be a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which would occur if the market value of our common stock held by non-affiliates was $700.0 million or more as of the last business day of the second fiscal quarter of such year or (iv) the date on which we have issued more than $1.0 billion in non-convertible debt securities during the prior three-year period.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to avail ourselves of this extended transition period and, as a result, we may adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-public companies instead of the dates required for other public companies.
We are also a “smaller reporting company” as defined in the Exchange Act. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies until the fiscal year following the determination that our voting and non-voting common stock held by non-affiliates is $250 million or more measured on the last business day of our second fiscal quarter, or our annual revenues are less than $100 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is $700 million or more measured on the last business day of our second fiscal quarter.
Corporate Information
We were formed in April 2021 as a Texas limited liability company under the name FibroBiologics, LLC, and converted to a Delaware corporation in December 2021 under the name Fibrobiologics, Inc. On April 12, 2023, we changed our name to FibroBiologics, Inc. Our principal executive offices are located at 455 E. Medical Center Blvd., Suite 300, Houston, Texas 77598. Our telephone number is (281) 671-5150 and our website address is www.fibrobiologics.com. Information contained on or that can be accessed through our website is neither a part of, nor incorporated by reference into, this prospectus, and you should not consider information on our website to be part of this prospectus. Our website address is included in this prospectus as an inactive textual reference only.
| 7 |
SUMMARY FINANCIAL AND OTHER DATA
The summary financial and other data set forth below should be read together with our financial statements and the related notes to those statements, as well as the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section of this prospectus. The statements of operations data for the years ended December 31, 2022 and 2021, and the statements of cash flows data for the years ended December 31, 2022 and 2021, have been derived from our audited financial statements included elsewhere in this prospectus. The statements of operations data for the nine months ended September 30, 2023 and 2022, the statements of cash flows data for the nine months ended September 30, 2023 and 2022, and the balance sheet data as of September 30, 2023, have been derived from our unaudited interim financial statements included elsewhere in this prospectus. The unaudited interim financial statements were prepared on a basis consistent with our audited financial statements and include in management’s opinion, all adjustments, consisting of normal recurring adjustments, that we consider necessary for a fair presentation of the financial information set forth in those statements. Our historical results are not necessarily indicative of the results that may be expected in any future period, and our interim results are not necessarily indicative of our expected results for the year ending December 31, 2023.
All share numbers and per share amounts in the tables below have been adjusted to reflect the Reverse Stock Split.
|
For the nine months ended September 30, |
For the years ended December 31, |
|||||||||||||||
| 2023 | 2022 | 2022 | 2021 | |||||||||||||
| (unaudited, in thousands, except shares and per share data) | (in thousands, except shares and per share data) | |||||||||||||||
| Statements of Operations Data: | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 1,595 | $ | 802 | $ | 1,147 | $ | 521 | ||||||||
| General, administrative and other | 4,814 | 2,361 | 3,320 | 1,057 | ||||||||||||
| Total operating expenses | 6,409 | 3,163 | 4,467 | 1,578 | ||||||||||||
| Loss from operations | (6,409 | ) | (3,163 | ) | (4,467 | ) | (1,578 | ) | ||||||||
| Other income/(loss) | (213 | ) | — | — | — | |||||||||||
| Interest expense | (146 | ) | (434 | ) | (654 | ) | (4 | ) | ||||||||
| Net loss | $ | (6,768 | ) | $ | (3,597 | ) | $ | (5,121 | ) | $ | (1,582 | ) | ||||
| Deemed dividend | (2,573 | ) | — | — | — | |||||||||||
| Net loss attributable to common stockholders | $ | (9,341 | ) | $ | (3,597 | ) | $ | (5,121 | ) | $ | (1,582 | ) | ||||
| Net loss per share, basic and diluted | $ | (.33 | ) | $ | (.13 | ) | $ | (.18 | ) | $ | N/A | |||||
| Weighted-average shares outstanding, basic and diluted | 28,230,842 | 28,230,842 | 28,230,842 | N/A | ||||||||||||
| Statements of Cash Flows Data: | ||||||||||||||||
| Net cash used in operating activities | $ | (4,800 | ) | $ | (2,893 | ) | $ | (4,066 | ) | $ | (1,410 | ) | ||||
| Net cash used in investing activities | $ | (493 | ) | $ | — | $ | — | $ | — | |||||||
| Net cash provided by financing activities | $ | 13,793 | $ | 3,775 | $ | 5,925 | $ | 1,817 | ||||||||
|
As of September 30, 2023 |
||||
| (unaudited, in thousands) | ||||
| Balance Sheet Data: | ||||
| Cash and cash equivalents | $ | 10,766 | ||
| Working capital¹ | $ | 9,600 | ||
| Total assets | $ | 13,299 | ||
| Total liabilities | $ | 2,761 | ||
| Total stockholders’ equity | $ | 10,538 | ||
¹ We define working capital as current assets less current liabilities.
| 8 |
An investment in our common stock involves a high degree of risk. You should carefully consider the following risks and uncertainties, together with all of the other information contained in this prospectus, including our financial statements and related notes appearing elsewhere in this prospectus, before deciding whether to invest in our common stock. The occurrence of one or more of the events or circumstances described in these risk factors, alone or in combination with other events or circumstances, may have a material adverse effect on our business, reputation, revenue, financial condition, results of operations and future prospects, in which event you could lose all or part of your investment. The risks and uncertainties described below are not intended to be exhaustive and are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations. This prospectus also contains forward-looking statements that involve risks and uncertainties. See “Cautionary Note Regarding Forward-Looking Statements.” Our actual results could differ materially and adversely from those anticipated in these forward-looking statements as a result of certain factors, including those described below.
Risks Related to Our Financial Condition and Capital Requirements
The successful development of biopharmaceutical products is highly uncertain.
Successful development of biopharmaceutical products is highly uncertain and is dependent on numerous factors, many of which are beyond our control. Product candidates that appear promising in the early phases of development may fail to reach the market for several reasons, including:
| ● | clinical trial results showing the product candidates to be less effective than expected (for example, a clinical trial could fail to meet its primary or key secondary endpoint(s)) or have an unacceptable safety or tolerability profile; |
| ● | failure to receive the necessary regulatory approvals or a delay in receiving such approvals, which, among other things, may be caused by patients who fail the trial screening process, slow enrollment in clinical trials, patients dropping out of trials, patients lost to follow-up, length of time to achieve trial endpoints, additional time requirements for data analysis or NDA preparation, discussions with the FDA, an FDA request for additional preclinical or clinical data or unexpected safety or manufacturing issues; |
| ● | preclinical study results showing the product candidate to be less effective than desired or to have harmful side effects; |
| ● | post-marketing approval requirements; or |
| ● | the proprietary rights of others and their competing products and technologies that may prevent our product candidates from being commercialized. |
The length of time necessary to complete clinical trials and submit an application for marketing approval for a final decision by a regulatory authority varies significantly from one product candidate to the next and from one country or jurisdiction to the next and may be difficult to predict.
Even if we are successful in obtaining marketing approval, commercial success of approved products may also depend in large part on the availability of coverage and adequate reimbursement from third-party payors, including government payors such as the Medicare and Medicaid programs and managed care organizations in the United States or country-specific governmental organizations in foreign countries, which may be affected by existing and future healthcare reform measures designed to reduce the cost of healthcare. Third-party payors could require us to conduct additional studies, including post-marketing studies related to the cost effectiveness of an approved product, to qualify for reimbursement, which could be costly and divert our resources. If government and other healthcare payors were to not provide coverage and adequate reimbursement for our products once approved, market acceptance and commercial success may be reduced.
| 9 |
In addition, if any of our product candidates receive marketing approval, we will be subject to significant regulatory obligations regarding the submission of safety and other post-marketing information and reports and registration, and will need to continue to comply (or ensure that any third-party providers comply) with cGMPs and good clinical practices, or GCPs, for any clinical trials that we conduct post-approval. In addition, there is always the risk that we, a regulatory authority or a third party might identify previously unknown problems with a product post-approval, such as adverse events of unanticipated severity or frequency. Compliance with these requirements is costly, and any failure to comply or other issues with our product candidates post-approval could adversely affect our business, financial condition and results of operations.
We have a limited operating history and none of our current product candidates have been approved for commercial sale, which may make it difficult for you to evaluate our current business and predict our future success and viability.
Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We are a clinical-stage cell therapy company with a limited operating history upon which you can evaluate our business and prospects. None of our current product candidates are approved for commercial sale and we have not generated any revenue from such product candidates. To date, we have devoted substantially all of our resources and efforts to organizing and staffing our company, business planning, executing partnerships, raising capital, discovering, identifying and developing potential product candidates, securing related intellectual property rights and conducting and planning preclinical studies and clinical trials of our product candidates. In relation to our current product candidates, we have not yet demonstrated our ability to successfully complete any Phase 3 clinical trials, obtain marketing approvals, manufacture a commercial-scale product or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. As a result, it may be more difficult for you to accurately predict our future success or viability than it could be if we had a longer operating history or a history of successfully developing and commercializing biopharmaceutical products.
In addition, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors and risks frequently experienced by clinical-stage biopharmaceutical companies in rapidly evolving fields. We also may need to transition from a company with a research focus to a company capable of supporting commercial activities. If we do not adequately address these risks and difficulties or successfully make such a transition, our business will suffer.
We have incurred significant net losses since inception, expect to continue to incur significant net losses for the foreseeable future, and may never achieve or maintain profitability.
We have incurred significant net losses since our inception, have not generated any revenue from product sales to date and have financed our operations principally through private financings. For the years ended December 31, 2022 and 2021, and the nine months ended September 30, 2023, we incurred net losses of $5.1 million, $1.6 million, and $6.8 million respectively. As of December 31, 2022, and September 30, 2023, we had an accumulated deficit of $7.9 million and $14.6 million, respectively. Our losses have resulted principally from expenses incurred in research and development of our product candidates and from management and administrative costs and other expenses that we have incurred while building our business infrastructure. We expect that it will be several years, if ever, before we have a commercialized product and generate revenue from product sales. Even if we succeed in receiving marketing approval for, and commercializing, one or more of our product candidates, we expect that we will continue to incur substantial research and development and other expenses as we discover, develop and market additional potential product candidates.
We expect to continue to incur significant losses for the foreseeable future, and we expect these losses to increase substantially if and as we:
| ● | advance the development of our lead product candidates through clinical development, and, if approved by the FDA, commercialization; |
| ● | advance our preclinical development programs into clinical development; |
| ● | incur manufacturing costs for cell production to supply our product candidates; |
| 10 |
| ● | seek regulatory approvals for any of our product candidates that successfully complete clinical trials; |
| ● | increase our research and development activities to identify and develop new product candidates; |
| ● | hire additional personnel; |
| ● | expand our operational, financial and management systems; |
| ● | meet the requirements and demands of being a public company; |
| ● | invest in further development to protect and expand our intellectual property; |
| ● | establish a sales, marketing, medical affairs and distribution infrastructure to commercialize any product candidates for which we may obtain marketing approval and intend to commercialize; and |
| ● | expand our manufacturing and develop our commercialization efforts. |
The net losses we incur may fluctuate significantly from period to period, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenue. Our prior losses and expected future losses have had and will continue to have an adverse effect on our working capital and our ability to achieve and maintain profitability.
Our ability to become and remain profitable depends on our ability to generate revenue or execute other business development arrangements. We do not expect to generate significant revenue, if any, unless and until we are able to obtain regulatory approval for, and successfully commercialize, one or more product candidates we are developing or may develop. Successful commercialization will require achievement of many key milestones, including demonstrating safety and efficacy in clinical trials, obtaining regulatory approval for these product candidates, manufacturing, marketing and selling those products for which we may obtain regulatory approval, satisfying any post-marketing requirements and obtaining reimbursement for our products from private insurance or government payors. Because of the uncertainties and risks associated with these activities, we are unable to accurately and precisely predict the timing and amount of revenues, the extent of any further losses or if or when we might achieve profitability.
We may never succeed in these activities and, even if we do, we may never generate revenues that are significant enough for us to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations. If we continue to incur losses as we have since our inception, investors may not receive any return on their investment and may lose their entire investment.
We will require substantial additional capital to finance our operations. If we are unable to raise such capital when needed, or on acceptable terms, we may be forced to delay, reduce and/or eliminate one or more of our research and drug development programs or future commercialization efforts.
Developing biopharmaceutical products, including conducting preclinical studies and clinical trials, is a very time-consuming, expensive and uncertain process that takes years to complete. Our operations have consumed substantial amounts of cash since inception, and we expect our expenses to increase in connection with our ongoing activities, particularly as we initiate and conduct clinical trials of, and seek marketing approval for our current product candidates and any future product candidates. Even if one or more of the product candidates that we develop is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate. Our expenses could increase beyond expectations if we are required by the FDA, the EMA or other comparable regulatory authorities to perform clinical trials or preclinical studies in addition to those that we currently anticipate. Other unanticipated costs may also arise. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to drug sales, marketing, manufacturing and distribution. Because the design and outcome of our anticipated clinical trials are highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of any product candidate we develop. Accordingly, we will need to obtain substantial additional funding in order to maintain our continuing operations.
| 11 |
As of September 30, 2023, we had approximately $10.8 million in cash and cash equivalents. Based on our current business plans, we believe that our existing capital will enable us to fund our operations through at least September 30, 2024. Our estimate as to how long we expect our existing capital to be able to continue to fund our operations is based on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect. Changing circumstances, some of which may be beyond our control, could cause us to consume capital significantly faster than we currently anticipate, and we may need to seek additional funds sooner than planned.
Our future funding requirements will depend on many factors, including, but not limited to:
| ● | the initiation, progress, timeline, cost and results of our clinical trials for our product candidates; |
| ● | the initiation, progress, timeline, cost and results of additional research and preclinical studies related to pipeline development and other research programs we initiate in the future; |
| ● | the cost and timing of manufacturing activities, including our planned manufacturing scale-up activities associated with our product candidates and other programs as we advance them through preclinical and clinical development through commercialization; |
| ● | the potential expansion of our current development programs to seek new indications; |
| ● | the outcome, timing and cost of meeting regulatory requirements established by the FDA and other comparable foreign regulatory authorities; |
| ● | the cost of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights, in-licensed or otherwise; |
| ● | the effect of competing technological and market developments; |
| ● | the payment of licensing fees, potential royalty payments and potential milestone payments; |
| ● | the cost of general operating expenses; |
| ● | the cost of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval in regions where we choose to commercialize our products on our own; and |
| ● | the costs of operating as a public company. |
Advancing the development of our product candidates will require a significant amount of capital. In order to fund all of the activities that are necessary to complete the development of our product candidates, we will be required to obtain further funding through equity offerings, debt financings, collaborations and licensing arrangements or other sources, which may dilute our stockholders or restrict our operating activities. Adequate additional funding may not be available to us on acceptable terms, or at all.
Our failure to raise capital as and when needed or on acceptable terms would have a negative impact on our financial condition and our ability to pursue our business strategy, and we may have to delay, reduce the scope of, suspend or eliminate one or more of our research-stage programs, clinical trials or future commercialization efforts, grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves, obtain funds through arrangement with collaborators on terms unfavorable to us or pursue merger or acquisition strategies, all of which could adversely affect the holdings or the rights of our stockholders.
| 12 |
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our product candidates on unfavorable terms to us.
We may seek additional capital through a variety of means, including through equity, debt financings, or other sources, including up-front payments and milestone payments from strategic collaborations. We may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences and anti-dilution protections that adversely affect your rights as a stockholder.
Such financing may also result in imposition of debt covenants, increased fixed payment obligations or other restrictions that may adversely affect our ability to conduct our business. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, or grant licenses on terms that are not favorable to us.
We are party to a share purchase agreement, dated November 12, 2021, with certain investors, or the Share Purchase Agreement, pursuant to which we may elect to issue and sell to such investors, and if so elected, such investors will be obligated to purchase, for a period commencing on the first day on which our common stock trade on a principal U.S. securities exchange and ending 60 months from such date, up to $100,000,000 worth of shares of our common stock, or the Aggregate Limit. The Share Purchase Agreement is contingent upon our achieving a public listing of our common stock. Pursuant to the agreement, we are required to pay the investors a commitment fee equal to 2% of the Aggregate Limit, payable in cash or shares of our common stock. The commitment fee is payable even if we do not utilize any drawdowns.
In addition, the agreement requires us to issue to the investors, on our public listing date, a warrant to purchase up to the number of shares of our common stock that is equal to 4% of our total equity interests outstanding immediately after the completion of our public listing, at a price per share equal to the lesser of (i) the public offering price per share (in the case of an initial public offering) or the closing bid price per share on the public listing date (in the case of a public listing other than an initial public offering) or (ii) the quotient obtained by dividing $700,000,000 by the total number of equity interests.
Our election to issue and sell to the investors, shares of our common stock pursuant to the Share Purchase Agreement, or the exercise of the warrant we will be obligated to issue upon consummation of this Direct Listing, will result in further dilution to our existing stockholders and investors who purchase shares of our common stock in this offering.
Risks Related to Development, Regulatory Approval and Commercialization
The regulatory approval processes of the FDA, the EMA and other comparable foreign regulatory authorities are lengthy, time consuming and inherently unpredictable. If we are ultimately unable to obtain regulatory approval for our product candidates, we will be unable to generate product revenue and our business will be substantially harmed.
We are not permitted to commercialize, market, promote or sell any product candidate in the United States without obtaining marketing approval from the FDA. Foreign regulatory authorities, such as the EMA, impose similar requirements. The time required to obtain approval by the FDA, the EMA and other comparable foreign regulatory authorities is unpredictable, typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the type, complexity and novelty of the product candidates involved. In addition, approval policies, regulations or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the approval or the decision not to approve an application. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other data. Even if we eventually complete clinical testing and receive approval of any regulatory filing for our product candidates, the FDA, the EMA and other comparable foreign regulatory authorities may approve our product candidates for a more limited indication or a narrower patient population than we originally requested. We have not submitted for, or obtained, regulatory approval for any product candidate, and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Further, development of our product candidates and/or regulatory approval may be delayed for reasons beyond our control.
Applications for our product candidates could fail to receive regulatory approval for many reasons, including the following:
| ● | the FDA, the EMA or other comparable foreign regulatory authorities may disagree with the design, implementation or results of our clinical trials; |
| ● | the FDA, the EMA or other comparable foreign regulatory authorities may determine that our product candidates are not safe and effective, only moderately effective or have undesirable or unintended side effects, toxicities or other characteristics that preclude our obtaining marketing approval or prevent or limit commercial use; |
| 13 |
| ● | the population studied in the clinical trial may not be sufficiently broad or representative to assure efficacy and safety in the full population for which we seek approval; |
| ● | the FDA, the EMA or other comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| ● | the data collected from clinical trials of our product candidates may not be sufficient to support the submission of a Biologics License Application, or BLA, or other submission or to obtain regulatory approval in the United States or elsewhere; |
| ● | we may be unable to demonstrate to the FDA, the EMA or other comparable foreign regulatory authorities that a product candidate’s risk-benefit ratio for its proposed indication is acceptable; |
| ● | the FDA, the EMA or other comparable foreign regulatory authorities may fail to approve our manufacturing processes, test procedures and specifications or facilities or those of third-party manufacturers with which we contract for clinical and commercial supplies; and |
| ● | the approval policies or regulations of the FDA, the EMA or other comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
This lengthy, uncertain approval process, as well as the unpredictability of the results of clinical trials, may result in our failing to obtain regulatory approval to market any of our product candidates, which would significantly harm our business, results of operations and prospects. In addition, the FDA, the EMA or comparable foreign regulatory authorities may change their policies, adopt additional regulations or revise existing regulations or take other actions, which may prevent or delay approval of our future product candidates under development on a timely basis. Such policy or regulatory changes could impose additional requirements upon us that could delay our ability to obtain approvals, increase the costs of compliance or restrict our ability to maintain any marketing authorizations we may have obtained.
We may encounter substantial delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
Before obtaining marketing approval from the FDA, the EMA or other comparable foreign regulatory authorities for the sale of our product candidates, we must complete preclinical development and extensive clinical trials to demonstrate the safety and efficacy of our product candidates. Clinical testing is expensive, difficult to design and implement, can take many years to complete and its ultimate outcome is uncertain. A failure of one or more clinical trials can occur at any stage of the process. The outcome of preclinical studies and early-stage clinical trials may not be predictive of the success of later clinical trials.
We do not know whether our future clinical trials will begin on time or enroll patients on time, or whether our ongoing and/or future clinical trials will be completed on schedule or at all. Clinical trials can be delayed for a variety of reasons, including delays related to:
| ● | the FDA, the EMA or other comparable foreign regulatory authorities disagreeing as to the design or implementation of our clinical trials; |
| ● | obtaining regulatory authorizations to commence a trial or reaching a consensus with regulatory authorities on trial design; |
| ● | any failure or delay in reaching an agreement with CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| ● | obtaining approval from one or more independent institutional review boards, or IRBs; |
| ● | IRBs refusing to approve, suspending or terminating the trial at an investigational site, precluding enrollment of additional subjects, or withdrawing their approval of the trial; |
| 14 |
| ● | delays in enrollment due to travel or quarantine policies, or other factors related pandemics or other events outside our control; |
| ● | changes to clinical trial protocol; |
| ● | clinical sites deviating from trial protocol or dropping out of a trial; |
| ● | manufacturing sufficient quantities of a product candidate or obtaining sufficient quantities of combination therapies for use in clinical trials; |
| ● | subjects failing to enroll or remain in our trial at the rate we expect, or failing to return for post- treatment follow-up; |
| ● | subjects choosing an alternative treatment for the indication for which we are developing our product candidates, or participating in competing clinical trials; |
| ● | lack of adequate funding to continue the clinical trial; |
| ● | subjects experiencing severe or unexpected drug-related adverse effects; |
| ● | occurrence of serious adverse events in trials of the same class of agents conducted by other companies; |
| ● | selection of clinical endpoints that require prolonged periods of clinical observation or analysis of the resulting data; |
| ● | a facility manufacturing our product candidates or any of their components being ordered by the FDA, the EMA or comparable foreign regulatory authorities to temporarily or permanently shut down due to violations of cGMP regulations or other applicable requirements, or infections or cross-contaminations of product candidates in the manufacturing process; |
| ● | any changes to our manufacturing process that may be necessary or desired; |
| ● | third-party clinical investigators losing the licenses or permits necessary to perform our clinical trials, not performing our clinical trials on our anticipated schedule or consistent with the clinical trial protocol, GCP or other regulatory requirements; |
| ● | third-party contractors not performing data collection or analysis in a timely or accurate manner; or |
| ● | third-party contractors becoming debarred or suspended or otherwise penalized by the FDA, the EMA or other government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or all of the data produced by such contractors in support of our marketing applications. |
Conducting clinical trials in foreign countries, as we may do for our product candidates, presents additional risks that may delay completion of our clinical trials. These risks include the failure of enrolled patients in foreign countries to adhere to clinical protocol as a result of differences in healthcare services or cultural customs, managing additional administrative burdens associated with foreign regulatory schemes, as well as political and economic risks relevant to such foreign countries.
| 15 |
Additionally, if the results of our clinical trials are inconclusive or if there are safety concerns or serious adverse events associated with our product candidates, we may:
| ● | be delayed in obtaining marketing approval, if at all; |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| ● | be required to perform additional clinical trials to support approval or be subject to additional post-marketing testing requirements; |
| ● | be subject to the addition of labeling statements, such as warnings or contraindications; |
| ● | be sued; or |
| ● | experience damage to our reputation. |
Our development costs will also increase if we experience delays in testing or obtaining marketing approvals. We do not know whether any of our preclinical studies or clinical trials will begin as planned, need to be restructured or be completed on schedule, if at all. Any delay in, or termination of, our clinical trials will delay the submission of a BLA to the FDA or similar applications with comparable foreign regulatory authorities and, ultimately, our ability to commercialize our product candidates, if approved, and generate product revenue. Even if our clinical trials are completed as planned, we cannot be certain that their results will support our claims for differentiation or the effectiveness or safety of our product candidate. The FDA has substantial discretion in the review and approval process and may disagree that our data support the claims we propose.
Moreover, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA, the EMA or other comparable foreign regulatory authorities. The FDA, the EMA or other comparable foreign regulatory authorities may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the study. The FDA, the EMA or other comparable foreign regulatory authorities may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA, the EMA or other comparable foreign regulatory authorities, as the case may be, and may ultimately lead to the denial of marketing approval of one or more of our product candidates.
If we experience delays in the completion of, or termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. Moreover, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues.
In addition, many of the factors that cause, or lead to, termination or suspension of, or a delay in the commencement or completion of, clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate. Any delays to our clinical trials that occur as a result could shorten any period during which we may have the exclusive right to commercialize our product candidates and our competitors may be able to bring products to market before we do, and the commercial viability of our product candidates could be significantly reduced. Any of these occurrences may harm our business, financial condition and prospects significantly.
| 16 |
The outcome of preclinical studies or early clinical trials may not be predictive of the success of later clinical trials, and the results of our clinical trials may not satisfy the requirements of the FDA, the EMA or other comparable foreign regulatory authorities.
Positive results from preclinical studies and early clinical trials do not mean that future clinical trials will be successful. Failure can occur at any time during the clinical trial process. We do not know whether any of our product candidates will perform in current or future clinical trials as they have performed in preclinical studies and early clinical trials. Product candidates in later-stage clinical trials may fail to demonstrate sufficient safety and efficacy to the satisfaction of the FDA, the EMA and other comparable foreign regulatory authorities despite having progressed through preclinical studies and early-stage clinical trials.
In some instances, there can be significant variability in safety and efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial protocols, differences in size and type of the patient populations, differences in and adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants. Patients treated with our product candidates may also be undergoing surgical, radiation and chemotherapy treatments and may be using other approved products or investigational new drugs, which can cause side effects or adverse events that are unrelated to our product candidate. As a result, assessments of efficacy can vary widely for a particular patient, and from patient to patient and site to site within a clinical trial. This subjectivity can increase the uncertainty of, and adversely impact, our clinical trial outcomes. We do not know whether any clinical trials we may conduct will demonstrate consistent or adequate efficacy and safety sufficient to obtain marketing approval to market our product candidates. Most product candidates that begin clinical trials are never approved by regulatory authorities for commercialization.
Additionally, some of our planned clinical trials may utilize an “open-label” trial design. An “open-label” clinical trial is one where both the patient and investigator know whether the patient is receiving either the investigational product candidate or an existing approved pharmaceutical or placebo. Most typically, open-label clinical trials test only the investigational product candidate and sometimes may do so at different dose levels. Open-label clinical trials are subject to various limitations that may exaggerate any therapeutic effect as patients in open-label clinical trials are aware when they are receiving treatment. Open-label clinical trials may be subject to a “patient bias” where patients perceive their symptoms to have improved merely due to their awareness of receiving an experimental treatment. In addition, open-label clinical trials may be subject to an “investigator bias” where those assessing and reviewing the physiological outcomes of the clinical trials are aware of which patients have received treatment and may interpret the information of the treated group more favorably given this knowledge. The results from an open-label trial may not be predictive of future clinical trial results with any of our product candidates for which we include an open-label clinical trial when studied in a controlled environment with a placebo or active control.
Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses and many companies that believed their product candidates performed satisfactorily in preclinical studies or clinical trials nonetheless failed to obtain FDA, EMA or comparable foreign regulatory authority approval. We cannot guarantee that the FDA, the EMA or comparable foreign regulatory authorities will interpret trial results as we do, and more trials could be required before we are able to submit applications seeking approval of our product candidates. This is particularly true for clinical trials in rare diseases, where the very small patient population makes it difficult to conduct two traditional, adequate and well-controlled studies, and therefore the FDA, the EMA or comparable foreign regulatory authorities are often required to exercise flexibility in approving therapies for such diseases. To the extent that the results of the trials are not satisfactory to the FDA, the EMA or comparable foreign regulatory authorities for support of a marketing application, we may be required to expend significant resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates. Even if regulatory approval is secured for any of our product candidates, the terms of such approval may limit the scope and use of our product candidate, which may also limit its commercial potential. Furthermore, the approval policies or regulations of the FDA, the EMA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval, which may lead to the FDA, the EMA or comparable foreign regulatory authorities delaying, limiting or denying approval of our product candidates.
| 17 |
Interim, topline and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose interim, preliminary or topline data from our preclinical studies or clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the interim, topline or preliminary results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Interim, preliminary and topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim, topline and preliminary data should be viewed with caution until the final data are available. Adverse differences between preliminary, topline or interim data and final data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure. If the interim, topline or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Our current or future product candidates may cause adverse events, toxicities or other undesirable side effects when used alone or in combination with other approved products or investigational new drugs that may result in a safety profile that could inhibit regulatory approval, prevent market acceptance, limit their commercial potential or result in significant negative consequences.
As is the case with biopharmaceuticals generally, it is likely that there may be side effects and adverse events associated with our product candidates’ use. Results of our clinical trials could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics. Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, the EMA or comparable foreign regulatory authorities. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
If our product candidates are associated with undesirable side effects or have unexpected characteristics in preclinical studies or clinical trials when used alone or in combination with other approved products or investigational new drugs we may need to interrupt, delay or abandon their development or limit development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Treatment-related side effects could also affect patient recruitment or the ability of enrolled subjects to complete the trial, or result in potential product liability claims. Any of these occurrences may prevent us from achieving or maintaining market acceptance of the affected product candidate and may harm our business, financial condition and prospects significantly.
Patients in our ongoing and planned clinical trials in the future may suffer significant adverse events or other side effects not observed in our preclinical studies or previous clinical trials. Some of our product candidates may be used as chronic therapies or be used in pediatric populations, for which safety concerns may be particularly scrutinized by regulatory agencies. In addition, if our product candidates are used in combination with other therapies, our product candidates may exacerbate adverse events associated with the therapy. Patients treated with our product candidates may also be undergoing surgical, radiation or chemotherapy treatments, which can cause side effects or adverse events that are unrelated to our product candidate but may still impact the success of our clinical trials. The inclusion of critically ill patients in our clinical trials may result in deaths or other adverse medical events due to other therapies or medications that such patients may be using or due to the gravity of such patients’ illnesses.
If significant adverse events or other side effects are observed in any of our current or future clinical trials, we may have difficulty recruiting patients to the clinical trials, patients may drop out of our trials, or we may be required to abandon the trials or our development efforts of that product candidate altogether. We, the FDA, the EMA, other comparable regulatory authorities or an IRB may suspend clinical trials of a product candidate at any time for various reasons, including a belief that subjects in such trials are being exposed to unacceptable health risks or adverse side effects.
| 18 |
Additionally, if any of our product candidates receives regulatory approval and becomes a product, and we or others later identify undesirable side effects caused by such product, a number of potentially significant negative consequences could result. For example, the FDA could require us to adopt a Risk Evaluation and Mitigation Strategy, or REMS, to ensure that the benefits of treatment with such product outweigh the risks for each potential patient, which may include, among other things, a communication plan to health care practitioners, patient education, extensive patient monitoring or distribution systems and processes that are highly controlled, restrictive and more costly than what is typical for the industry. We or our collaborators may also be required to adopt a REMS or engage in similar actions, such as patient education, certification of health care professionals or specific monitoring, if we or others later identify undesirable side effects caused by any product that we develop alone or with collaborators. Other potentially significant negative consequences include that:
| ● | we may be forced to suspend marketing of that product, or decide to remove the product from the marketplace; |
| ● | regulatory authorities may withdraw or change their approvals of that product; |
| ● | regulatory authorities may require additional warnings on the label or limit access of that product to selective specialized centers with additional safety reporting and with requirements that patients be geographically close to these centers for all or part of their treatment; |
| ● | we may be required to create a medication guide outlining the risks of the product for patients, or to conduct post-marketing studies; |
| ● | we may be required to change the way the product is administered; |
| ● | we could be subject to fines, injunctions, or the imposition of criminal or civil penalties, or be sued and held liable for harm caused to subjects or patients; and |
| ● | the product may become less competitive, and our reputation may suffer. |
Any of these events could diminish the usage or otherwise limit the commercial success of our product candidates and prevent us from achieving or maintaining market acceptance of the affected product candidate, if approved by applicable regulatory authorities.
Even if approved, our product candidates may not achieve adequate market acceptance among physicians, patients, healthcare payors and others in the medical community necessary for commercial success.
Even if our product candidates receive regulatory approval and become a product, they may not gain adequate market acceptance among physicians, patients, healthcare payors and others in the medical community. The degree of market acceptance of any of our approved product candidates will depend on a number of factors, including:
| ● | the efficacy and safety profile as demonstrated in clinical trials compared to alternative treatments; |
| ● | the timing of market introduction of the product as well as competitive products; |
| ● | the clinical indications for which the product is approved; |
| ● | restrictions on the use of our product, such as boxed warnings or contraindications in labeling, or a REMS, if any, which may not be required of alternative treatments and competitor products; |
| ● | the potential and perceived advantages of products over alternative treatments; |
| ● | the cost of treatment in relation to alternative treatments; |
| 19 |
| ● | the availability of coverage and adequate reimbursement, as well as pricing, by third-party payors, including government authorities; |
| ● | relative convenience and ease of administration; |
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| ● | the effectiveness of sales and marketing efforts; |
| ● | unfavorable publicity relating to our products or similar approved products or product candidates in development by third parties; and |
| ● | the approval of other new therapies for the same indications. |
If any of our product candidates is approved but does not achieve an adequate level of acceptance by physicians, hospitals, healthcare payors and patients, we may not generate or derive sufficient revenue from that product candidate and our financial results could be negatively impacted.
Our refrigerated product candidates require specific storage, handling and administration at the clinical sites.
Our refrigerated drug product candidates must be stored at low temperatures in specialized refrigerated containers until immediately prior to use. For administration, the drug product container must be carefully removed from storage, warmed to room temperature and inverted to place cells into suspension prior to drawing the product into syringes. The handling, warming and administration of the cell therapy product must be performed according to specific instructions. Failure to correctly handle the product, follow the instructions for warming and administration and/or failure to administer the product within the specified period post-warming could negatively impact the efficacy and or safety of the product.
Because cell therapy is novel and the regulatory landscape that governs any cell therapy product candidates we may develop is rigorous, complex, uncertain and subject to change, we cannot predict the time and cost of obtaining regulatory approval, if we receive it at all, for any product candidates we may develop. At the moment, only a small number of cell therapy products have been approved in the United States and the European Union.
The regulatory requirements that will govern any novel cell therapy product candidates we develop are not entirely clear and are subject to change. Within the broader genetic medicine field, very few therapeutic products have received marketing authorization from the FDA or the EMA. Even with respect to more established products that fit into the categories of gene therapies or cell therapies, the regulatory landscape is still developing. Regulatory requirements governing cell therapy products have changed frequently and will likely continue to change in the future. Moreover, there is substantial overlap in those responsible for regulation of existing cell therapy products. For example, in the United States, the FDA has established the Office of Tissues and Advanced Therapies within its Center for Biologics Evaluation and Research to consolidate the review of cell therapy and related products. Although the FDA has approved other cell-based therapies, there is no assurance that these previous approvals will affect the FDA’s review of our product candidates.
Our cell therapy product candidates will need to meet safety and efficacy standards applicable to any new biologic under the regulatory framework administered by the FDA. In addition to FDA oversight and oversight by IRBs, under the National Institutes of Health Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules, or NIH Guidelines, cell therapy clinical trials are also subject to review and oversight by an Institutional Biosafety Committee, or IBC, a local institutional committee that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment. While the NIH Guidelines are not mandatory unless the research in question is being conducted at or sponsored by institutions receiving National Institutes of Health, or NIH, funding of recombinant or synthetic nucleic acid molecule research, many companies and other institutions not otherwise subject to the NIH Guidelines voluntarily follow them. Although the FDA decides whether individual cell therapy protocols may proceed, the review process and determinations of other reviewing bodies can impede or delay the initiation of a clinical trial, even if the FDA has reviewed the trial and approved its initiation.
| 20 |
The same applies in the European Union. The EMA’s Committee for Advanced Therapies, or CAT, is responsible for assessing the quality, safety, and efficacy of advanced-therapy medicinal products. Advanced-therapy medicinal products include cell therapy medicines, somatic-cell therapy medicines and tissue-engineered medicines. The role of the CAT is to prepare a draft opinion on an application for marketing authorization for a cell therapy medicinal candidate that is submitted to the EMA. In the European Union, the development and evaluation of a cell therapy product must be considered in the context of the relevant EU guidelines. The EMA may issue new guidelines concerning the development and marketing authorization for cell therapy products and require that we comply with these new guidelines. As a result, the procedures and standards applied to cell therapy products may be applied to any cell therapy product candidate we may develop, but that remains uncertain at this point.
Adverse developments in preclinical studies or clinical trials conducted by others in the field of cell therapy and cell regulation products may cause the FDA, the EMA and other regulatory bodies to revise the requirements for approval of any product candidates we may develop or limit the use of products utilizing cell therapy technologies, either of which could harm our business. In addition, the clinical trial requirements of the FDA, the EMA and other regulatory authorities and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty, and intended use and market of the potential products. The regulatory approval process for product candidates such as ours can be more expensive and take longer than for other, better known, or more extensively studied pharmaceutical or other product candidates. Further, as we are developing novel potential treatments for diseases in which, in some cases, there is little clinical experience with potential new endpoints and methodologies, there is heightened risk that the FDA, the EMA or other regulatory bodies may not consider the clinical trial endpoints to provide clinically meaningful results, and the resulting clinical data and results may be more difficult to analyze. In addition, we may not be able to identify or develop appropriate animal disease models to enable or support planned clinical development. Any natural history studies that we may conduct or rely upon in our clinical development may not be accepted by the FDA, the EMA or other regulatory authorities. Regulatory agencies administering existing or future regulations or legislation may not allow production and marketing of products utilizing cell therapy technology in a timely manner or under technically or commercially feasible conditions. In addition, regulatory action or private litigation could result in expenses, delays or other impediments to our research programs or the commercialization of resulting products. Further, approvals by one regulatory agency may not be indicative of what other regulatory agencies may require for approval.
The regulatory review committees and advisory groups described above and the new guidelines they promulgate may lengthen the regulatory review process, require us to perform additional preclinical studies or clinical trials, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of these product candidates, or lead to significant post-approval limitations or restrictions. As we advance our research programs and develop future product candidates, we will be required to consult with these regulatory and advisory groups and to comply with applicable guidelines. If we fail to do so, we may be required to delay or discontinue development of any product candidates we identify and develop. These additional processes may result in a review and approval process that is longer than we otherwise would have expected. Delays as a result of an increased or lengthier regulatory approval process or further restrictions on the development of our product candidates can be costly and could negatively impact our ability to complete clinical trials and commercialize our current and future product candidates in a timely manner, if at all.
We may be unable to obtain U.S. or foreign regulatory approvals and, as a result, may be unable to commercialize our product candidates.
Our product candidates are subject to extensive governmental regulations relating to, among other things, research, testing, development, manufacturing, safety, efficacy, approval, recordkeeping, reporting, labeling, storage, packaging, advertising and promotion, pricing, marketing and distribution of drugs. Rigorous preclinical studies and clinical trials and an extensive regulatory approval process must be successfully completed in the United States and in many foreign jurisdictions before a new drug can be marketed. Satisfaction of these and other regulatory requirements is costly, time-consuming, uncertain and subject to unanticipated delays. We cannot provide any assurance that any product candidate we may develop will progress through required clinical testing and obtain the regulatory approvals necessary for us to begin selling them.
| 21 |
We have not conducted, managed or completed large-scale or pivotal clinical trials nor managed the regulatory approval process with the FDA, the EMA or any other regulatory authority with respect to our current product candidates. The time required to obtain approvals from the FDA and other regulatory authorities is unpredictable and requires successful completion of extensive clinical trials which typically takes many years, depending upon the type, complexity and novelty of the product candidate. The standards that the FDA and its foreign counterparts use when evaluating clinical trial data can and often changes during drug development, which makes it difficult to predict with any certainty how they will be applied. We may also encounter unexpected delays or increased costs due to new government regulations, including future legislation or administrative action, or changes in FDA policy during the period of drug development, clinical trials and FDA regulatory review.
Any delay or failure in seeking or obtaining required approvals would have a material and adverse effect on our ability to generate revenue from the particular product candidate for which we are developing and seeking approval. Furthermore, any regulatory approval to market a product candidate may be subject to significant limitations on the approved uses or indications for which we may market the product candidate or the labeling or other restrictions. In addition, the FDA has the authority to require a REMS as part of approving an NDA or BLA, or after approval, which may impose further requirements or restrictions on the distribution or use of an approved product candidate. These requirements or restrictions might include limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. These limitations and restrictions may significantly limit the size of the market for the product candidate and affect reimbursement by third-party payors.
We are also subject to numerous foreign regulatory requirements governing, among other things, the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process varies among countries, and generally includes all of the risks associated with FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Moreover, the time required to obtain approval may differ from that required to obtain FDA approval.
We may develop our current and future product candidates in combination with other therapies, which exposes us to additional risks, and certain of our product candidates are regulated as combination products.
We may develop our current and future product candidates in combination with one or more other approved or unapproved therapies to treat skin and connective tissue diseases or other diseases. We may also develop certain product candidates as biologic/drug combination products. Additional time may be required to obtain regulatory approval for our product candidates because they are combination products. Our product candidates that are biologic/drug combination products require coordination within the FDA and similar foreign regulatory agencies for review of their biologic and drug components. Although the FDA and similar foreign regulatory agencies have systems in place for the review and approval of combination products such as ours, we may experience delays in the development and commercialization of our product candidates due to regulatory timing constraints and uncertainties in the product development and approval process.
In addition, even if any product candidate we develop were to receive marketing approval or be commercialized for use in combination with other existing therapies, we would continue to be subject to the risks that the FDA, the EMA or comparable foreign regulatory authorities could revoke approval of the therapy used in combination with our product or that safety, efficacy, manufacturing or supply issues could arise with any of those existing therapies. If the therapies we use in combination with our product candidates are replaced as the standard of care for the indications we choose for any of our product candidates, the FDA, the EMA or comparable foreign regulatory authorities may require us to conduct additional clinical trials. The occurrence of any of these risks could result in our own product candidates, if approved, being removed from the market or being less successful commercially.
We also may choose to evaluate our current product candidates or any future product candidates in combination with one or more therapies that have not yet been approved for marketing by the FDA, the EMA or comparable foreign regulatory authorities. We will not be able to market and sell our product candidates we develop in combination with an unapproved therapy for a combination indication if that unapproved therapy does not ultimately obtain marketing approval either alone or in combination with our product candidate. In addition, unapproved therapies face the same risks described with respect to our product candidates currently in development and clinical trials, including the potential for serious adverse effects, delay in their clinical trials and lack of FDA approval.
If the FDA, the EMA or comparable foreign regulatory authorities do not approve these other products or revoke their approval of, or if safety, efficacy, quality, manufacturing or supply issues arise with, the products we choose to evaluate in combination with our product candidates we develop, we may be unable to obtain approval of or market such combination therapy.
| 22 |
Any product candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, each as amended, or collectively, the ACA, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of their product. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement BPCIA may be fully adopted by the FDA, any such processes could have an adverse effect on the future commercial prospects for our biological products.
There is a risk that any of our product candidates approved as a biological product under a BLA would not qualify for the 12-year period of exclusivity or that this exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of litigation. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. If competitors are able to obtain marketing approval for biosimilars referencing our candidates, if approved, our products may become subject to competition from such biosimilars, with the attendant competitive pressure and potential adverse consequences.
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs, therapeutic platforms and product candidates that we identify for specific indications. As a result, we may forego or delay pursuit of opportunities with other therapeutic platforms or product candidates or for other indications that later prove to have greater commercial potential or a greater likelihood of success. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs, therapeutic platforms and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights.
Risks Related to Our Business
Our company has limited experience in designing clinical trials and may experience delays or unexpected difficulties in obtaining regulatory approval for our current and future product candidates.
We have limited experience in designing clinical trials and may be unable to design and execute a clinical trial to support marketing approval. We cannot be certain that our planned clinical trials or any future clinical trials will be successful. It is possible that the FDA may refuse to accept any or all of our planned BLAs for substantive review or may conclude after review of our data that our application is insufficient to obtain regulatory approval for any product candidates. If the FDA does not approve any of our planned BLAs, it may require that we conduct additional costly clinical trials, preclinical studies or manufacturing validation studies before it will reconsider our applications. Depending on the extent of these or any other FDA-required studies, approval of any BLA or other application that we submit may be significantly delayed, possibly for several years, or may require us to expend more resources than we have available. Any failure or delay in obtaining regulatory approvals would prevent us from commercializing our product candidates, generating revenues and achieving and sustaining profitability. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the FDA to approve any BLA or other application that we submit. If any of these outcomes occur, we may be forced to abandon the development of our product candidates, which would materially adversely affect our business and could potentially cause us to cease operations. We face similar risks for our applications in foreign jurisdictions.
| 23 |
We intend to identify and develop novel cell therapy product candidates, which makes it difficult to predict the time, cost and potential success of product candidate development.
Our strategy is to identify, develop and commercialize cell therapy product candidates using our proprietary fibroblast technology, which involves collecting skin biopsies from donor patients, isolating cells and expanding them in culture. Our future success depends on the successful development of these novel therapeutic approaches. To date, no fibroblast therapy products have been approved. In addition, there have been a few number of clinical trials involving fibroblasts as compared to other, more conventional forms of therapy.
The sizes of the markets for our product candidates are estimates, and these markets may be smaller than estimated.
The estimates in this prospectus of the annual addressable markets for our product candidates are based on a number of third-party estimates. While we believe the assumptions and the data underlying the estimates are reasonable, these assumptions and estimates may not be correct and the conditions supporting the assumptions or estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors. As a result, the estimates of the annual addressable market for our product candidates may prove to be incorrect.
Our long-term prospects depend in part upon discovering, developing and commercializing additional product candidates, which may fail in development or suffer delays that adversely affect their commercial viability.
Our future operating results are dependent on our ability to successfully discover, develop, obtain regulatory approval for and commercialize product candidates beyond those we currently have in clinical development. A product candidate can unexpectedly fail at any stage of preclinical or clinical development. The historical failure rate for product candidates is high due to risks relating to safety, efficacy, clinical execution, changing standards of medical care and other unpredictable variables. The results from preclinical studies or early clinical trials of a product candidate may not be predictive of the results that will be obtained in later stage clinical trials of the product candidate.
The success of other product candidates we may develop will depend on many factors, including the following:
| ● | generating sufficient data to support the initiation or continuation of clinical trials; |
| ● | obtaining regulatory permission to initiate clinical trials; |
| ● | contracting with the necessary parties to conduct clinical trials; |
| ● | successful enrollment of patients in, and the completion of, clinical trials on a timely basis; |
| ● | the timely manufacture of sufficient quantities of the product candidate and other key materials needed for use in clinical trials; and |
| ● | adverse events in the clinical trials. |
Even if we successfully advance any other product candidates into clinical development, their success will be subject to all of the clinical, regulatory and commercial risks described elsewhere in this “Risk Factors” section. Accordingly, we cannot assure you that we will ever be able to discover, develop, obtain regulatory approval of, commercialize or generate significant revenue from our product candidates.
| 24 |
We have never commercialized a fibroblast cell-based therapy product candidate before and may lack the necessary expertise, personnel and resources to successfully commercialize any product candidates, if approved, on our own or together with suitable collaborators.
We have never commercialized a fibroblast cell-based therapy product candidate, and we currently have no sales force, marketing or distribution capabilities. To achieve commercial success for our current product candidates, which we may license to others, we will rely on the assistance and guidance of those collaborators. For any approved product candidates for which we retain commercialization rights, we will have to develop our own sales, marketing and supply organization or outsource these activities to a third party.
Factors that may affect our ability to commercialize our product candidates on our own include recruiting and retaining adequate numbers of effective sales and marketing personnel, obtaining access to or persuading adequate numbers of physicians to prescribe our product candidates and other unforeseen costs associated with creating an independent sales and marketing organization. Developing a sales and marketing organization will be expensive and time-consuming and could delay the launch of our product candidates, if approved. We may not be able to build an effective sales and marketing organization. If we are unable to build our own distribution and marketing capabilities or to find suitable partners for the commercialization of our product candidates, we may not generate revenues from them or be able to reach or sustain profitability.
We face significant competition, and if our competitors develop and market technologies or products more rapidly than we do or that are more effective, safer or less expensive than the product candidates we develop, our commercial opportunities will be negatively impacted.
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary and novel products and product candidates. Our competitors have developed, are developing or may develop products, product candidates and processes competitive with our product candidates. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. We believe that a significant number of products are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may attempt to develop product candidates. See “Business—Competition” for additional details. In addition, our products may need to compete with off-label drugs used by physicians to treat the indications for which we seek approval. This may make it difficult for us to replace existing therapies with our products.
Many current and potential competitors have significantly greater financial, manufacturing, marketing, drug development, technical and human resources and commercial expertise than we do. Large pharmaceutical and biotechnology companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing biotechnology products. These companies also have significantly greater research and marketing capabilities than we do and may also have products that have been approved or are in late stages of development, and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical and biotechnology companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies, as well as in acquiring technologies complementary to, or necessary for, our programs. As a result, our competitors may succeed in obtaining approval from the FDA, the EMA or other comparable foreign regulatory authorities or in discovering, developing and commercializing products in our field before we do.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe effects, are more convenient, have a broader label, are marketed more effectively, are reimbursed or are less expensive than any product candidates that we may develop. Our competitors also may obtain marketing approval from the FDA, the EMA or other comparable foreign regulatory authorities for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. Even if the product candidates we develop achieve marketing approval, they may be priced at a significant premium over competitive products if any have been approved by then, resulting in reduced competitiveness. Technological advances or products developed by our competitors may render our technologies or product candidates obsolete, less competitive or uneconomical. If we are unable to compete effectively, our opportunity to generate revenue from the sale of any products we may develop, if approved, could be adversely affected.
| 25 |
We will be a “controlled company” within the meaning of the Nasdaq Stock Market Rules after this offering because our insiders will beneficially own more than 50% of the voting power of our outstanding voting securities.
Upon completion of this offering, our founder and Chief Executive Officer, Pete O’Heeron, will collectively beneficially own approximately 59% of the voting power of our outstanding voting securities and we will be a “controlled company” within the meaning of the listing rules of The Nasdaq Stock Market LLC. We may rely on certain exemptions from corporate governance rules, including an exemption from the rule that a majority of our board of directors must be independent directors. Although we currently do not intend to rely on the “controlled company” exemption under the Nasdaq listing rules, we could elect to rely on this exemption in the future. In the event that we elected to rely on the “controlled company” exemption, a majority of the members of our board of directors might not be independent directors, and our nominating and corporate governance and compensation committees might not consist entirely of independent directors. Our status as a controlled company could cause our shares of common stock to be less attractive to certain investors or otherwise harm our trading price. As a result, you would not have the same protection afforded to shareholders of companies that are subject to these corporate governance requirements.
We will incur increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives. We will be subject to financial reporting and other requirements for which our accounting and other management systems and resources may not be adequately prepared.
As a public company, and particularly after we are no longer an emerging growth company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, the federal securities laws, including the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, and rules and regulations subsequently implemented by the SEC and Nasdaq have imposed various requirements on public companies, including requirements to file annual, quarterly, and event driven reports with respect to their business and financial condition, and to establish and maintain effective disclosure and financial controls and corporate governance practices. These rules and regulations will increase our legal and financial compliance costs, make certain activities more time-consuming and costly, and require our management and other personnel to devote a substantial amount of time to compliance initiatives.
Despite our best efforts, we may not be able to produce reliable financial statements or file such financial statements as part of a periodic report in a timely manner with the SEC or comply with Nasdaq listing requirements. We also expect that these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance.
Pursuant to Section 404 of the Sarbanes-Oxley Act, we will be required to furnish a report by our management on our internal control over financial reporting, including an attestation report on internal control over financial reporting issued by our independent registered public accounting firm, beginning with the first full year after we become a public company. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 of the Sarbanes-Oxley Act, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. We will need to continue to dedicate internal resources, potentially engage outside consultants, adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that neither we nor our independent registered public accounting firm will be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404 of the Sarbanes-Oxley Act. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements. We could also become subject to investigations by the SEC or other regulatory authorities, which could require additional financial and management resources.
As a public company, we will also be required to maintain disclosure controls and procedures. Disclosure controls and procedures means our controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the rules and forms of the SEC. We do not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent or detect all errors and all fraud. We believe a control system, no matter how well-designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Due to the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, have been detected. The design of any system of controls is based in part on certain assumptions about the likelihood of future events, and any design may not succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
We have identified a material weakness in our internal control over financial reporting due to lack of segregation of duties. Failure to maintain effective internal control over financial reporting could cause our investors to lose confidence in us and adversely affect the market price of our common stock. If our internal controls over financial reporting are not effective, we may not be able to accurately report our financial results or prevent fraud.
During the preparation of our financial statements for the fiscal year ended December 31, 2022, our management identified a material weakness in our internal control over financial reporting due to a lack of segregation of duties. A material weakness is defined as a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
Specifically, our management identified a deficiency in our internal controls within the financial reporting function that resulted from a lack of segregation of duties for the period of time covered by our financial statements prior to our Chief Financial Officer joining us in June 2022 when all financial functions were handled by a single individual.
With the addition of our Chief Financial Officer and the changes made to our accounting and financial reporting processes and internal controls during the last half of fiscal year 2022, we have strengthened our internal controls and will continue to evaluate segregation of duties and take initiatives to improve our internal controls over financial reporting as we grow. However, the implementation of these initiatives may not fully address the material weakness in our internal control over financial reporting and we cannot assure you that we will not identify other material weaknesses or deficiencies, which could negatively impact our results of operations in future periods.
| 26 |
Risks Relating to Our Dependence on Third Parties
We rely, and expect to continue to rely, on third parties, including independent clinical investigators and CROs, to conduct certain aspects of our preclinical studies and clinical trials. If these third parties do not successfully carry out their contractual duties, comply with applicable regulatory requirements or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be adversely harmed.
We have relied upon and plan to continue to rely upon third parties, including independent clinical investigators and third-party CROs, to conduct certain aspects of our preclinical studies and clinical trials and to monitor and manage data for our ongoing preclinical and clinical programs. We rely on these parties for execution of our preclinical studies and clinical trials, and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies and trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on these third parties does not relieve us of our regulatory responsibilities. We, our third-party contractors and CROs are required to comply with GCP requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for all of our product candidates in clinical development. Regulatory authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of these third parties or our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, the EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP regulations. In addition, our clinical trials must be conducted with products manufactured under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process. Moreover, our business may be adversely affected if any of these third parties violates federal or state fraud and abuse or false claims laws and regulations or healthcare privacy and security laws.
Further, there is no guarantee that any such CROs, investigators or other third parties on which we rely will devote adequate time and resources to our development activities or perform as contractually required. These third parties may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other product development activities, which could affect their performance on our behalf. If independent investigators or CROs fail to devote sufficient resources to the development of our product candidates, or if CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. Consequently, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed or halted entirely.
Our CROs have the right to terminate their agreements with us in the event of an uncured material breach. In addition, some of our CROs have an ability to terminate their respective agreements with us if it can be reasonably demonstrated that the safety of the subjects participating in our clinical trials warrants such termination, if we make a general assignment for the benefit of our creditors or if we are liquidated.
If any of our relationships with these third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms. Switching or adding additional CROs involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Additionally, CROs may lack the capacity to absorb higher workloads or take on additional capacity to support our needs. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
| 27 |
If we decide to establish additional collaborations but are not able to establish those collaborations on commercially reasonable terms, we may have to alter our development and commercialization plans.
Our product candidate development programs and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. We may continue to seek to selectively form collaborations to expand our capabilities, potentially accelerate research and development activities and provide for commercialization activities by third parties. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing stockholders, or disrupt our management and business.
We would face significant competition in seeking appropriate collaborators and the negotiation process is time-consuming and complex. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA, the EMA or comparable foreign regulatory authorities, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of intellectual property and industry and market conditions generally. The potential collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such collaboration could be more attractive than the one with us for our product candidate. Further, we may not be successful in our efforts to establish a collaboration or other alternative arrangements for future product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view them as having the requisite potential to demonstrate safety and efficacy.
In addition, mergers among large biopharmaceutical companies may result in a reduced number of potential future collaborators. Even if we are successful in entering into a collaboration, the terms and conditions of that collaboration may restrict us from entering into future agreements on certain terms with potential collaborators.
If and when we seek to enter into collaborations, we may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of a product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
In the future we may enter into collaborations with third parties for the development and commercialization of product candidates. If those collaborations are not successful, we may not be able to capitalize on the market potential of these product candidates.
We may in the future seek third-party collaborators for the development and commercialization of one or more of our product candidates. Our likely collaborators for any future collaboration arrangements include large and mid-size biopharmaceutical companies, regional and national biopharmaceutical companies and biotechnology companies. We have and will likely have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities and efforts to successfully perform the functions assigned to them in these arrangements. Collaborations involving our product candidates could pose numerous risks to us, including the following:
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations and may not perform their obligations as expected; |
| 28 |
| ● | collaborators may deemphasize or not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborators’ strategic focus, including as a result of a sale or disposition of a business unit or development function, or available funding or external factors such as an acquisition that diverts resources or creates competing priorities; |
| ● | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
| ● | a collaborator with marketing and distribution rights to multiple products may not commit sufficient resources to the marketing and distribution of our product candidates relative to other products; |
| ● | collaborators may not properly obtain, maintain, defend or enforce our intellectual property rights or may use our proprietary information and intellectual property in such a way as to invite litigation or other intellectual property-related proceedings that could jeopardize or invalidate our proprietary information and intellectual property or expose us to potential litigation or other intellectual property-related proceedings; |
| ● | disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our product candidates or that result in costly litigation or arbitration that diverts management attention and resources; |
| ● | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates; |
| ● | collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all; and |
| ● | if a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product candidate development or commercialization program could be delayed, diminished or terminated. |
Our employees, independent contractors, consultants, commercial collaborators, principal investigators, CROs, suppliers and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have an adverse effect on our results of operations.
We are exposed to the risk that our employees, independent contractors, consultants, commercial collaborators, principal investigators, CROs, suppliers and vendors may engage in misconduct or other improper activities. Misconduct by these parties could include failures to comply with FDA regulations, provide accurate information to the FDA, comply with federal and state health care fraud and abuse laws and regulations, accurately report financial information or data or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct by these parties could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter misconduct by these parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant penalties, including civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, integrity oversight and reporting obligations, contractual damages, reputational harm, diminished profits and future earnings and the curtailment or restructuring of our operations.
| 29 |
Risks Related to Manufacturing
Manufacturing cell therapy products is complex and subject to both human and systemic risks. Our third-party manufacturers or we may encounter difficulties in production and sourcing and may be subject to variations and supply constraints of critical components. If we or any of our third-party manufacturers encounter such difficulties, our ability to supply our product candidates for clinical trials or our products for patients, if approved, could be delayed or prevented.
The manufacture of biologic cell therapy product candidates, and products, if approved, is complex and requires significant expertise and capital investment, including developing advanced manufacturing techniques and process controls. Manufacturers of biologic products often encounter difficulties in production and sourcing, particularly in scaling up or out, validating the production process, and assuring high reliability of the manufacturing processes (including the absence of contamination), in light of variations and supply constraints of critical components. These problems include logistics and shipping, difficulties with production costs and yields, quality control, including consistency, stability, purity, and efficacy of the product, product testing, operator error, and availability of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. Furthermore, if contaminants are discovered in our supply of our product candidates or in the manufacturing facilities, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. We cannot assure you that any stability, purity, and efficacy failures, deficiencies, or other issues relating to manufacturing our product candidates will not occur in the future.
Additionally, our product candidates are derived from cells collected from humans. Such cells may vary in type and quality as the donors may vary in age, medical history and many other factors. We have strict specifications for donor cell material and our product candidates. The donor cell material variability may exceed our manufacturing process capability or deviate from the specified ranges and result in failure in the production of the cell therapy product, lower quality batches, or even require adjustments to the specifications approved by authorities. The donor cell material may also be variable in factors that we currently may not be able to detect with the analytical methods used or may not know how to measure. We may also discover failures with the material after production. As a result, we may not be able to deliver the quality and consistency of our cell therapy products that we need or may need to re-collect cell material which can increase costs and/or cause delay, adversely impact patient outcomes and otherwise harm our clinical trials, reputation, business and prospects.
We may fail to manage the logistics of collecting and shipping patient material to the manufacturing site, shipping the product candidate back to the relevant parties, and experiencing delays or shortages of certain clinical or commercial-grade supplies and components. Logistical and shipment delays and problems caused by us, our vendors, or other factors not in our control, including business interruptions, global supply chain issues, and weather, could prevent or delay the delivery of product candidates to patients. Additionally, we have to maintain a complex chain of identity and chain of custody with respect to donor material as it moves to the manufacturing facility, through the manufacturing processes, and ultimately to a patient. Failure to maintain a chain of identity and chain of custody could result in patient death, loss of product, or regulatory action.
The transfer or production of our cell banks to a contract development manufacturing organization may fail and result in delays, additional costs, or technical failure.
We currently purchase our cell therapy product candidates from a contract development and manufacturing organization, or CDMO. We are in the process of contracting with a CDMO, for the transfer of our experimental cell bank to produce our master cell bank, working cell bank and our fibroblast cell-based product candidates to enable clinical trials. If the transfer of our experimental cell bank to the CDMO is not successful, we may encounter delays, additional costs, or technical failure of one or more of our product candidates. Similarly, if the CDMO is unable to produce from the experimental cell bank our master cell bank, working cell bank and our fibroblast cell-based product candidates to enable clinical trials, we may encounter delays, additional costs, or technical failure of one or more of our product candidates.
Changes in the methods of product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates proceed through preclinical studies to late-stage clinical trials towards potential marketing approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, formulation, materials and processes, are altered along the way in an effort to optimize processes and product characteristics. Such alterations can also occur due to changes in manufacturers. Such changes carry the risk that they will not achieve their intended objectives. Any such changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with product candidates produced using the modified manufacturing methods, materials and processes. Such changes may also require additional testing, FDA notification or FDA approval. This could delay the completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one or more clinical trials beyond those we currently anticipate, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commercialize our product candidates if approved. In addition, we may be required to make significant changes to our upstream and downstream processes across our pipeline, which could delay the development of future product candidates.
| 30 |
If we or our third-party manufacturers use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Research and development activities involve the controlled use of potentially hazardous substances, including chemical and biological materials, by us and our third-party manufacturers. We currently outsource all manufacturing to third parties. Still, we and our manufacturers are subject to federal, state and local laws and regulations in the United States governing the use, manufacture, storage, handling and disposal of medical and hazardous materials. Although we believe that our manufacturers’ procedures for using, handling, storing and disposing of these materials comply with legally prescribed standards, we cannot completely eliminate the risk of contamination or injury resulting from medical or hazardous materials. As a result of any such contamination or injury, we may incur liability, or local, city, state or federal authorities may curtail the use of these materials and interrupt our business operations. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources. We do not currently have any insurance for liabilities arising from medical or hazardous materials. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts, which could harm our business, prospects, financial condition or results of operations.
We rely on third parties for our manufacturing process and may, in the future, depend on third-party manufacturers for our product candidates, and this increases the risk related to the timely and sufficient production of our product candidates.
We do not have complete control over all aspects of the manufacturing process of, and are dependent on, our contract manufacturing partners for compliance with cGMP regulations for manufacturing our cell therapy product candidates. Third-party manufacturers may be unable to comply with cGMP regulations or similar regulatory requirements outside the United States. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA, the EMA or others, they will not be able to secure and/or maintain marketing approval for their manufacturing facilities. In addition, we do not have control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA, the EMA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain marketing approval for or market our product candidates, if approved. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates and harm our business and results of operations. Furthermore, the raw materials for our product candidates may be sourced, in some cases, from a single-source supplier. If we were to experience an unexpected loss of supply of any of our product candidates or any of our future product candidates for any reason, whether as a result of manufacturing, supply, or storage issues or otherwise, we could experience delays, disruptions, suspensions or terminations of, or be required to restart or repeat, any pending or ongoing clinical trials.
We currently rely on third-party manufacturers to produce our product candidates for use in development and commercialization under the guidance of members of our organization. In the event that we or any of our third-party manufacturers fail to comply with such requirements or to perform with certain requirements in relation to quality, timing, or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to enter into an agreement with another third party, which we may not be able to do on commercially reasonable terms, if at all. In particular, any replacement of our third-party manufacturers could require significant effort and expertise because there may be a limited number of qualified replacements. In some cases, the technical skills or technology required to manufacture our product candidates may be unique or proprietary to us or the third-party manufacturer. We may have difficulty transferring such skills or technology to another third party, and a feasible alternative may not exist. In addition, certain of our product candidates and our own proprietary methods have never been produced or implemented outside of our company. Therefore, we may experience delays in our development programs if we attempt to establish new third-party manufacturing arrangements for these product candidates or methods. These factors would increase our reliance on such manufacturers or require us to obtain a license from such manufacturers in order to have another third party manufacture our product candidates. If we are required to or voluntarily stop manufacturing our product candidates for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines and that the product produced is equivalent to that produced in our facility. The delays associated with the verification of a new manufacturer and equivalent product could negatively affect our ability to develop product candidates in a timely manner or within budget.
| 31 |
Our or a third party’s failure to execute our manufacturing requirements, do so on commercially reasonable terms and timelines, and comply with cGMP requirements could adversely affect our business in a number of ways, including:
| ● | inability to meet our product specifications and quality requirements consistently; |
| ● | inability to initiate or continue clinical trials of our product candidates under development; |
| ● | delays in submitting regulatory applications or receiving marketing approvals for our product candidates, if at all; |
| ● | inability to commercialize any product candidates that receive marketing approval on a timely basis; |
| ● | loss of the cooperation of future collaborators; |
| ● | subjecting third-party manufacturing facilities or our manufacturing facilities to additional inspections by regulatory authorities; |
| ● | requirements to cease development or to recall batches of our product candidates; and |
| ● | in the event of approval to market and commercialize our product candidates, an inability to meet commercial demands for our product candidates or any future product candidates. |
Any contamination or interruption in our manufacturing processes, shortages of raw materials, or failure of our suppliers to deliver necessary components could result in delays in our clinical development or marketing schedules.
Given the nature of cell therapy manufacturing, there is a risk of contamination. Any contamination could adversely affect our ability to produce product candidates on schedule and could, therefore, harm our results of operations and cause reputational damage. Additionally, although our cell therapies are tested for contamination prior to release if a contaminated product candidate was administered to a patient, it could result in harm to the patient. Some of the raw materials required in our manufacturing process are derived from biological sources. Such raw materials are difficult to procure and may be subject to contamination or recall. A material shortage, contamination, recall, or restriction on the use of biologically derived substances in the manufacture of our product candidates could adversely impact or disrupt the commercial manufacturing or the production of clinical material, which could adversely affect our development timelines and our business, financial condition, results of operations and prospects.
Risks Related to Legal and Regulatory Compliance Matters
Our relationships with healthcare professionals, clinical investigators, CROs and third-party payors in connection with our current and future business activities may be subject to federal and state healthcare fraud and abuse laws, false claims laws, transparency laws, government price reporting, and health information privacy and security laws, which could expose us to, among other things, criminal sanctions, civil penalties, contractual damages, exclusion from governmental healthcare programs, reputational harm, administrative burdens and diminished profits and future earnings.
Healthcare providers and third-party payors play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our current and future arrangements with healthcare professionals, clinical investigators, CROs, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our product candidates for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for the purchase, lease, order, arrangement, or recommendation of any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it to have committed a violation. Violations are subject to civil fines and criminal penalties for each violation, plus up to three times the remuneration involved, imprisonment, and exclusion from government healthcare programs. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act or federal civil money penalties; |
| 32 |
| ● | the federal civil and criminal false claims laws and civil monetary penalty laws, such as the federal False Claims Act, which impose criminal and civil penalties and authorize civil whistleblower or qui tam actions, against individuals or entities for, among other things: knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent; knowingly making, using or causing to be made or used, a false statement of record material to a false or fraudulent claim or obligation to pay or transmit money or property to the federal government or knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay money to the federal government. Manufacturers can be held liable under the federal False Claims Act even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. The federal False Claims Act also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the federal False Claims Act and to share in any monetary recovery; |
| ● | the Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits, among other things, a person from knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false, fictitious, or fraudulent statements or representations in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● | HIPAA, as further amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, including the Final Omnibus Rule published in January 2013, which impose certain requirements on certain covered healthcare providers, health plans and healthcare clearinghouses, as well as their respective business associates, independent contractors or agents of covered entities, that perform services for them that involve the use, creation, maintenance, receipt or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, there are additional federal, state and non-U.S. laws which govern the privacy and security of health and other personal information in certain circumstances to which we may be subject and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; |
| ● | federal government price reporting laws, which require manufacturers to calculate and report complex pricing metrics in an accurate and timely manner to government programs; |
| 33 |
| ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| ● | The ACA, including the provision commonly referred to as the Physician Payments Sunshine Act and its implementing regulations, which require applicable manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually to the U.S. Centers for Medicare & Medicaid Services, or CMS, within the U.S. Department of Health and Human Services, information related to payments or other transfers of value made to physicians, nurse practitioners, certified nurse anesthetists, physician assistants, clinical nurse specialists, and certified nurse midwives as well as teaching hospitals and to disclose ownership and investment interests held by physicians and their immediate family members; and |
| ● | many state laws that govern the privacy of personal information in specified circumstances. For example, in California, the California Consumer Privacy Act, or the CCPA, which went into effect on January 1, 2020, establishes a new privacy framework for covered businesses by creating an expanded definition of personal information, establishing new data privacy rights for consumers in the State of California, imposing special rules on the sale of personal information, and creating a new and potentially severe statutory damages framework for violations of the CCPA and for businesses that fail to implement reasonable security procedures and practices to prevent data breaches. While clinical trial data and information governed by HIPAA are currently exempt from the CCPA, other personal information collection practices may be subject to the CCPA and possible changes to the CCPA may broaden its scope. |
Additionally, we are subject to state and foreign equivalents of each of the healthcare laws and regulations described above, among others, some of which may be broader in scope and may apply regardless of the payor. Many U.S. states have adopted laws similar to the federal Anti-Kickback Statute and False Claims Act, and may apply to our business practices, including, but not limited to, research, distribution, sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental payors, including private insurers. In addition, some states have passed laws that require biopharmaceutical companies to comply with the April 2003 Office of Inspector General Compliance Program Guidance for Pharmaceutical Manufacturers and/or the Pharmaceutical Research and Manufacturers of America’s Code on Interactions with Healthcare Professionals. Several states also impose other marketing restrictions or require biopharmaceutical companies to make marketing or price disclosures to the state and require the registration of biopharmaceutical sales representatives. Privacy and data protection laws from outside of the United States, including, for example, the European Union General Data Protection Regulation and the UK Data Protection Act 2018, or, collectively, the GDPR, also govern the privacy and security of personal information, including health information in some circumstances, and many of these laws differ from each other in significant ways, thus complicating compliance efforts. In addition, in the United States, there are a number of states that have enacted laws that govern the privacy and security of personal information, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. There are ambiguities as to what is required to comply with these state requirements and if we fail to comply with an applicable state law requirement we could be subject to penalties.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Ensuring business arrangements comply with applicable healthcare and privacy laws, as well as responding to possible investigations by government authorities, can be time and resource-consuming and can divert a company’s attention from the business.
| 34 |
Ensuring that our internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant penalties, including administrative, civil and criminal penalties, damages, fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment, reputational harm and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. Further, defending against any such actions can be costly and time consuming, and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If any of the physicians or other providers or entities with whom we do, or expect to do, business is found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs and imprisonment. If any of the above occur, our ability to operate our business and our results of operations could be adversely affected.
We may be or become subject to evolving global data protection laws and regulations, which may require us to incur substantial compliance costs, and any failure or perceived failure by us to comply with such laws and regulations may harm our business and operations.
The global data protection landscape is rapidly evolving, and we may be or become subject to or affected by numerous federal, state and foreign laws and regulations, as well as regulatory guidance, governing the collection, use, disclosure, transfer, security and processing of personal data, such as information that we collect about participants and healthcare providers in connection with clinical trials. Implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future, which may create uncertainty in our business, affect our ability to operate in certain jurisdictions or to collect, store, transfer use and share personal data, result in liability or impose additional compliance or other costs on us. Any failure or perceived failure by us to comply with federal, state, or foreign laws or self-regulatory standards could result in negative publicity, diversion of management time and effort and proceedings against us by governmental entities or others. For example, states, such as California, Virginia, Colorado, Utah and Connecticut have recently enacted consumer privacy laws that grant rights to data subjects and places privacy and security obligations on entities handling personal data of consumers or households. Some observers note that the CCPA and similar legislation could mark the beginning of a trend toward more stringent privacy legislation in the United States, which could increase our potential liability and adversely affect our business.
In addition to our operations in the United States, which may be subject to healthcare and other laws relating to the privacy and security of health information and other personal information, we may seek to conduct clinical trials in the United Kingdom or the European Economic Area, or the EEA, and may become subject to additional European data privacy laws, regulations and guidelines. We will be subject to the data protection laws of the European Union and United Kingdom in relation to personal data we collect from these territories. These laws impose additional obligations and risk upon our business, including substantial expenses and changes to business operations that are required to comply with these laws. The withdrawal of the United Kingdom from the European Union, or Brexit, and the subsequent separation of the data protection regimes of these territories mean we are required to comply with separate data protection laws in the European Union and United Kingdom, which may lead to additional compliance costs and could increase our overall risk.
The GDPR, which deals with the processing of personal data and on the free movement of such data, imposes a broad range of strict requirements, including requirements relating to having lawful bases for processing personal data and transferring such information outside the EEA/UK, including to the United States, providing details to those individuals regarding the processing of their personal data, keeping personal data secure, having data processing agreements with third parties who process personal data, responding to individuals’ requests to exercise their rights in respect of their personal data, reporting security breaches involving personal data to the competent national data protection authority and affected individuals, appointing data protection officers, conducting data protection impact assessments and record-keeping.
| 35 |
The GDPR imposes strict rules on the transfer of personal data out of the EEA/UK to countries not regarded by European Commission and the United Kingdom government as providing adequate protection, or the third countries, including the United States. These transfers are prohibited unless an appropriate safeguard specified by data protection laws is implemented, such as the Standard Contractual Clauses, or the SCCs, approved by the European Commission, or a derogation applies. The UK has published its own transfer mechanism, the International Data Transfer Agreement and International Data Transfer Addendum, which enables transfers from the UK and has implemented a similar Transfer Equivalence Test. The international transfer obligations under the EU and UK data protection regimes require effort and cost and may result in us needing to make strategic considerations around where EEA/UK personal data is located and which service providers we utilize for the processing of EEA/UK personal data, particularly as the enforcement around GDPR international transfer compliance obligations is currently unclear. The UK Government has also now introduced a Data Protection and Digital Information Bill, or the UK Bill, into the UK legislative process with the intention for this bill to reform the UK’s data protection regime. If passed, the final version of the UK Bill may have the effect of further altering the similarities between the UK and EU data protection regime. This may lead to additional compliance costs and could increase our overall risk.
We cannot assure you that any efforts to comply with any obligations under European privacy laws will be sufficient. If we are investigated by a European data protection authority, we may face fines and other penalties. Any such investigation or charges by European data protection authorities could have a negative effect on our reputation and materially harm our business.
Our business entails a significant risk of product liability and if we are unable to obtain sufficient insurance coverage such inability could have an adverse effect on our business and financial condition.
Our business exposes us to significant product liability risks inherent in the development, testing, manufacturing and marketing of therapeutic treatments. Product liability claims could delay or prevent completion of our development programs. If we succeed in marketing product candidates, such claims could result in an FDA, EMA or other regulatory authority investigation of the safety and effectiveness of our product candidates, our manufacturing processes and facilities or our marketing programs. FDA, EMA or other regulatory authority investigations could potentially lead to a recall of our product candidates or more serious enforcement action, limitations on the approved indications for which they may be used or suspension or withdrawal of approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our product candidates, if approved, injury to our reputation, costs to defend the related litigation, a diversion of management’s time and our resources and substantial monetary awards to trial participants or patients. We currently have product liability insurance that we believe is appropriate for our stage of development and may need to obtain higher levels prior to marketing any of our product candidates, if approved. Any insurance we have or may obtain may not provide sufficient coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims that could have an adverse effect on our business and financial condition.
Any product candidates we develop may become subject to unfavorable third-party coverage and reimbursement practices, as well as pricing regulations.
The availability and extent of coverage and adequate reimbursement by third-party payors, including government health administration authorities, private health coverage insurers, managed care organizations and other third-party payors is essential for most patients to be able to afford expensive treatments. Sales of any of our product candidates that receive marketing approval will depend substantially, both in the United States and internationally, on the extent to which the costs of our product candidates will be covered and reimbursed by third-party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize an adequate return on our investment. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not successfully commercialize any product candidate for which we obtain marketing approval. For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult because of the higher prices often associated with such drugs.
| 36 |
There is significant uncertainty related to third-party payor coverage and reimbursement of newly approved products. In the United States, no uniform policy of coverage and reimbursement for products exists among third-party payors and coverage and reimbursement levels for products can differ significantly from payor to payor. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. However, one third-party payor’s determination to provide coverage for a product candidate does not assure that other payors will also provide coverage for the product candidate. As a result, the coverage determination process is often time-consuming and costly. This process will require us to provide scientific and clinical support for the use of our product candidates to each third-party payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Factors payors consider in determining reimbursement are based on whether the product is: (i) a covered benefit under its health plan; (ii) safe, effective and medically necessary; (iii) appropriate for the specific patient; (iv) cost-effective; and (v) neither experimental nor investigational.
Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. Further, such payors are increasingly challenging the price, examining the medical necessity and reviewing the cost effectiveness of medical product candidates. There may be especially significant delays in obtaining coverage and reimbursement for newly approved drugs. Third-party payors may limit coverage to specific product candidates on an approved list, known as a formulary, which might not include all FDA-approved drugs for a particular indication. We may need to conduct expensive pharmacoeconomic studies to demonstrate the medical necessity and cost effectiveness of our product candidates. Nonetheless, our product candidates may not be considered medically necessary or cost effective. We cannot be sure that coverage and reimbursement will be available for any product candidate that we commercialize and, if reimbursement is available, what the level of reimbursement will be.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost containment initiatives in Europe, Canada and other countries has and will continue to put pressure on the pricing and usage of therapeutics such as our product candidates. In many countries, particularly the countries of the European Union, medical product prices are subject to varying price control mechanisms as part of national health systems. In these countries, pricing negotiations with governmental authorities can take considerable time after a product receives marketing approval. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. In general, product prices under such systems are substantially lower than in the United States. Other countries allow companies to fix their own prices for products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our product candidates may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits.
If we are unable to establish or sustain coverage and adequate reimbursement for any future product candidates from third-party payors, the adoption of those product candidates and sales revenue will be adversely affected, which, in turn, could adversely affect the ability to market or sell those product candidates, if approved. Coverage policies and third-party payor reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more product candidates for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
We face potential liability related to the privacy of health information we obtain from clinical trials sponsored by us.
Most healthcare providers, including research institutions from which we obtain patient health information, are subject to privacy and security regulations promulgated under HIPAA, as amended by the HITECH. We are not currently classified as a covered entity or business associate under HIPAA and thus are not directly subject to its requirements or penalties. However, any person may be prosecuted under HIPAA’s criminal provisions either directly or under aiding-and-abetting or conspiracy principles. Consequently, depending on the facts and circumstances, we could face substantial criminal penalties if we knowingly receive individually identifiable health information from a HIPAA-covered healthcare provider or research institution that has not satisfied HIPAA’s requirements for disclosure of individually identifiable health information. In addition, we may maintain sensitive personally identifiable information, including health information, that we receive throughout the clinical trial process, in the course of our research collaborations, and directly from individuals (or their healthcare providers) who enroll in our patient assistance programs. As such, we may be subject to state laws requiring notification of affected individuals and state regulators in the event of a breach of personal information, which is a broader class of information than the health information protected by HIPAA.
| 37 |
Furthermore, certain health privacy laws, data breach notification laws, consumer protection laws and genetic testing laws may apply directly to our operations and/or those of our collaborators and may impose restrictions on our collection, use and dissemination of individuals’ health information. Patients about whom we or our collaborators obtain health information, as well as the providers who share this information with us, may have statutory or contractual rights that limit our ability to use and disclose the information. We may be required to expend significant capital and other resources to ensure ongoing compliance with applicable privacy and data security laws. Claims that we have violated individuals’ privacy rights or breached our contractual obligations, even if we are not found liable, could be expensive and time consuming to defend and could result in adverse publicity that could harm our business.
If we or third-party contract manufacturing organizations, CROs or other contractors or consultants fail to comply with applicable federal, state or local regulatory requirements, we could be subject to a range of regulatory actions that could affect our or our contractors’ ability to develop and commercialize our product candidates and could harm or prevent sales of any affected product candidates that we are able to commercialize, or could substantially increase the costs and expenses of developing, commercializing and marketing our product candidates. Any threatened or actual government enforcement action could also generate adverse publicity and require that we devote substantial resources that could otherwise be used in other aspects of our business. Increasing use of social media could give rise to liability, breaches of data security or reputational damage.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of hazardous and flammable materials, including chemicals and biological materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or commercialization efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
The FDA, the EMA and other comparable foreign regulatory authorities may not accept data from trials conducted in locations outside of their jurisdiction.
We may choose to conduct international clinical trials in the future. The acceptance of study data by the FDA, the EMA or other comparable foreign regulatory authority from clinical trials conducted outside of their respective jurisdictions may be subject to certain conditions. In cases where data from foreign clinical trials are intended to serve as the basis for marketing approval in the United States, the FDA will generally not approve the application on the basis of foreign data alone unless (i) the data are applicable to the United States population and United States medical practice; (ii) the trials are performed by clinical investigators of recognized competence and pursuant to current GCP requirements; and (iii) the FDA is able to validate the data through an on-site inspection or other appropriate mean. Additionally, the FDA’s clinical trial requirements, including the adequacy of the patient population studied and statistical powering, must be met. In addition, such foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are conducted. There can be no assurance that the FDA, the EMA or any applicable foreign regulatory authority will accept data from trials conducted outside of its applicable jurisdiction. If the FDA, the EMA or any applicable foreign regulatory authority does not accept such data, it would result in the need for additional trials, which would be costly and time-consuming and delay aspects of our business plan, and which may result in our product candidates not receiving approval for commercialization in the applicable jurisdiction.
| 38 |
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction. For example, even if the FDA or the EMA grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion and reimbursement of the product candidate in those countries. However, a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional preclinical studies or clinical trials as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our product candidates is also subject to approval.
Obtaining foreign regulatory approvals and establishing and maintaining compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our product candidates in certain countries. If we or any future collaborator fail to comply with the regulatory requirements in international markets or fail to receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of our product candidates will be harmed.
Even if our product candidates receive regulatory approval and become products, they will be subject to significant post-marketing regulatory requirements and oversight.
Any regulatory approvals that we may receive for our product candidates will require the submission of reports to regulatory authorities and surveillance to monitor the safety and efficacy of the products, may contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, and may include burdensome post-approval study or risk management requirements. For example, the FDA may require a REMS in order to approve our product candidates, which could entail requirements for a medication guide, physician training and communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA, the EMA or foreign regulatory authorities approve our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for our product candidates will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as on-going compliance with cGMPs and GCP for any clinical trials that we conduct post-approval. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic, unannounced inspections by the FDA and other regulatory authorities for compliance with cGMP regulations and standards. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facilities where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. In addition, failure to comply with FDA, the EMA or other comparable foreign regulatory requirements may subject our company to administrative or judicially imposed sanctions, including:
| ● | delays in or the rejection of product approvals; |
| ● | restrictions on our ability to conduct clinical trials, including full or partial clinical holds on ongoing or planned trials; |
| 39 |
| ● | restrictions on the products, manufacturers or manufacturing process; |
| ● | warning or untitled letters; |
| ● | civil and criminal penalties; |
| ● | injunctions; |
| ● | suspension or withdrawal of regulatory approvals; |
| ● | product seizures, detentions or import bans; |
| ● | voluntary or mandatory product recalls and publicity requirements; |
| ● | total or partial suspension of production; and |
| ● | imposition of restrictions on operations, including costly new manufacturing requirements. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenue and could require us to expend significant time and resources in response and could generate negative publicity.
The FDA’s and other regulatory authorities’ policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, and we may not achieve or sustain profitability.
We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad. For example, certain policies of the current U.S. administration may impact our business and industry. Namely, the previous U.S. administration took several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine regulatory and oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. It is difficult to predict whether or how these executive actions, including the Executive Orders, will be implemented, or whether they will be rescinded or replaced under the new U.S. administration. If these executive actions impose constraints on FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses.
If any of our product candidates are approved and we are found to have improperly promoted off-label uses of those products, we may become subject to significant liability. The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA, the EMA or such other regulatory agencies as reflected in the product’s approved labeling. If we receive marketing approval for a product candidate, physicians may nevertheless prescribe it to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability. The U.S. federal government has levied large civil and criminal fines against companies for alleged improper promotion of off-label use and has enjoined several companies from engaging in off-label promotion. The government has also required companies to enter into consent decrees or imposed permanent injunctions under which specified promotional conduct is changed or curtailed. If we cannot successfully manage the promotion of our product candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business and financial condition.
| 40 |
We may face difficulties from changes to current regulations and future legislation.
Existing regulatory policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
For example, the ACA substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. biopharmaceutical industry. Other legislative changes have been proposed and adopted in the United States since the ACA was enacted. These changes include aggregate reductions to Medicare payments and may result in additional reductions in Medicare and other healthcare funding, all of which could have a material adverse effect on customers for our product candidates, if approved, and accordingly, our financial operations.
There have also been several changes and challenges to the 340B drug pricing program, which imposes ceilings on prices that drug manufacturers can charge for medications sold to certain health care facilities. It is unclear how these developments could affect covered hospitals who might purchase our future product candidate and affect the rates we may charge such facilities for our approved product candidates in the future, if any.
Moreover, there has been heightened governmental scrutiny in recent years over the manner in which drug manufacturers set prices for their marketed products, which has resulted in several U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products. The U.S. Congress has indicated that it will continue to seek new legislative measures to control drug costs.
Further, on May 30, 2018, the Right to Try Act was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational new product candidates that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a drug manufacturer to make its products available to eligible patients as a result of the Right to Try Act.
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product candidates. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our product candidates.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for biotechnology products. We cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
Risks Related to Our Intellectual Property
Our success depends on our ability to protect our intellectual property and our proprietary technologies.
Our commercial success depends in part on our ability to obtain and maintain patent protection and trade secret protection for our product candidates, proprietary technologies and their uses to operate without infringing the proprietary rights of others. If we or our licensors are unable to protect our intellectual property rights or if our intellectual property rights are inadequate for our technology or our product candidates, our competitive position could be harmed. We and our licensors generally seek to protect our proprietary position by filing patent applications in the United States and abroad related to our product candidates, proprietary technologies and their uses that are important to our business. Our in-licensed patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless, and until, patents issue from such applications, and then only to the extent the issued claims cover the technology. There can be no assurance that our in-licensed patent applications will result in patents being issued or that issued patents will afford sufficient protection against competitors with similar technology, nor can there be any assurance that the patents if issued will not be infringed, designed around, invalidated or rendered unenforceable by third parties. Even issued patents may later be found invalid or unenforceable or may be modified or revoked in proceedings instituted by third parties before various patent offices or in courts. The degree of future protection for our and our licensors’ proprietary rights is uncertain. Only limited protection may be available and may not adequately protect our or our licensors’ rights or permit us or our licensors to gain or keep any competitive advantage. These uncertainties and/or limitations in our and our licensors’ ability to properly protect the intellectual property rights relating to our product candidates could have a material adverse effect on our financial condition and results of operations.
| 41 |
Although we may in-license issued patents in the United States and foreign countries, we cannot be certain that the claims in our other in-licensed U.S. pending patent applications, corresponding international patent applications and patent applications in certain foreign countries will be considered patentable by the United States Patent and Trademark Office, or USPTO, courts in the United States or by the patent offices and courts in foreign countries, nor can we be certain that the claims in our in-licensed issued patents will not be found invalid or unenforceable if challenged.
The patent application process is subject to numerous risks and uncertainties, and there can be no assurance that we or our licensors or any of our potential future collaborators will be successful in protecting our product candidates by obtaining and defending patents. These risks and uncertainties include the following:
| ● | the USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process, the noncompliance with which can result in abandonment or lapse of a patent or patent application, and partial or complete loss of patent rights in the relevant jurisdiction; |
| ● | patent applications may not result in any patents being issued; |
| ● | patents may be challenged, invalidated, modified, revoked, circumvented, found to be unenforceable or otherwise may not provide any competitive advantage; |
| ● | the degree and range of protection any issued patents will afford us against competitors, including whether third parties will find ways to invalidate or otherwise circumvent our patents; |
| ● | whether others will apply for or obtain patents claiming aspects similar to those covered by our patents and patent applications; |
| ● | whether the patent applications that we own or in-license will result in issued patents with claims that cover our product candidates or uses thereof in the United States or in other foreign countries; |
| ● | our competitors, many of whom have substantially greater resources than we or our licensors do and many of whom have made significant investments in competing technologies, may seek or may have already obtained patents that will limit, interfere with or block our ability to make, use and sell our product candidates; |
| ● | there may be significant pressure on the U.S. government and international governmental bodies to limit the scope of patent protection both inside and outside the United States for disease treatments that prove successful, as a matter of public policy regarding worldwide health concerns; and |
| ● | countries other than the United States may have patent laws less favorable to patentees than those upheld by U.S. courts, allowing foreign competitors a better opportunity to create, develop and market competing products. |
| 42 |
The patent prosecution process is also expensive and time-consuming, and we or our licensors may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner or in all jurisdictions where protection may be commercially advantageous. It is also possible that we or our licensors may not identify patentable aspects of our research and development output before it is too late to obtain patent protection. Moreover, in some circumstances, we do not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, directed to technology that we license, including those from our licensors and from third parties. We also may require the cooperation of our licensors in order to enforce the licensed patent rights, and such cooperation may not be provided. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. We cannot be certain that patent prosecution and maintenance activities by our licensors have been or will be conducted in compliance with applicable laws and regulations, which may affect the validity and enforceability of such patents or any patents that may issue from such applications. If they fail to do so, this could cause us to lose rights in any applicable intellectual property that we in-license, and as a result our ability to develop and commercialize products or product candidates may be adversely affected and we may be unable to prevent competitors from making, using and selling competing products.
Composition of matter patents for biological and pharmaceutical products such as cell therapy product candidates often provide a strong form of intellectual property protection for those types of products, as such patents provide protection without regard to any method of use. We cannot be certain, however, that the claims in our pending patent applications covering the composition of matter of our product candidates will be considered patentable by the USPTO, or by patent offices in foreign countries, or that the claims in any of our issued patents will be considered valid and enforceable by courts in the United States or foreign countries. Method of use patents protect the use of a product for the specified method. This type of patent does not prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside the scope of the patented method. Moreover, even if competitors do not actively promote their product for our targeted indications, physicians may prescribe these products “off-label” for those uses that are covered by our method of use patents. Although off-label prescriptions may infringe or contribute to the infringement of method of use patents, the practice is common and such infringement is difficult to prevent or prosecute.
In addition, although we enter into non-disclosure and confidentiality agreements with parties who have access to patentable aspects of our research and development output, such as our employees, outside scientific collaborators, CROs, third-party manufacturers, consultants, advisors, licensors and other third parties, any of these parties may breach such agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection.
If the scope of any patent protection our licensors obtain is not sufficiently broad, or if our licensors lose any of the patent protection we license, our ability to prevent our competitors from commercializing similar or identical product candidates would be adversely affected.
The patent position of biopharmaceutical companies generally is highly uncertain, involves complex legal and factual questions, and has been the subject of much litigation in recent years. As a result, the existence, issuance, scope, validity, enforceability and commercial value of our in-licensed patent rights are highly uncertain. Our pending and future in-licensed patent applications may not result in patents being issued that protect our product candidates or that effectively prevent others from commercializing competitive product candidates.
Moreover, the scope of claims in a patent application can be significantly reduced before any claims in a patent is issued, and claim scope can be reinterpreted after issuance. Even if patent applications we license currently or in the future issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with any competitive advantage. Any patents that we license may be challenged or circumvented by third parties or may be narrowed or invalidated as a result of challenges by third parties. Consequently, we do not know whether our product candidates will be protectable or remain protected by valid and enforceable patents. Our competitors or other third parties may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner, which could materially adversely affect our business, financial condition, results of operations and prospects.
| 43 |
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our licensed-in patents may not cover our product candidates or may be challenged in the courts or patent offices in the United States and abroad. We may be subject to a third party pre-issuance submission of prior art to the USPTO, or become involved in opposition, derivation, revocation, reexamination, post-grant review, or PGR, and inter partes review, or IPR, or other similar proceedings in the USPTO or foreign patent offices challenging our patent rights. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity of our in-licensed patents, for example, we cannot be certain that there is no invalidating prior art, of which we or our licensors and the patent examiner were unaware during prosecution. There is no assurance that all potentially relevant prior art relating to our in-licensed patents and patent applications or those of our licensors has been found. There is also no assurance that there is not prior art of which we or licensors are aware, but which we do not believe affects the validity or enforceability of a claim in our patents and patent applications or those of our licensors, which may, nonetheless, ultimately be found to affect the validity or enforceability of a claim. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate or render unenforceable, our in-licensed patent rights, allow third parties to commercialize our product candidates and compete directly with us, without payment to us. Such loss of licensed patent rights, loss of exclusivity or in patent claims being narrowed, invalidated or held unenforceable could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our product candidates. Such proceedings also may result in substantial cost and require significant time from our scientists and management, even if the eventual outcome is favorable to us. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a third-party patent, which might adversely affect our ability to develop and market our product candidates.
We cannot guarantee that any of our patent searches or analyses, including the identification of relevant patents, the scope of patent claims or the expiration of relevant patents, are complete or thorough, nor can we be certain that we have identified each and every third-party patent and pending application in the United States and abroad that is relevant to or necessary for the commercialization of our product candidates in any jurisdiction. The scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be incorrect, which may negatively impact our ability to market our products. We may incorrectly determine that our products are not covered by a third-party patent or may incorrectly predict whether a third-party’s pending application will issue with claims of relevant scope. Our determination of the expiration date of any patent in the United States or abroad that we consider relevant may be incorrect, which may negatively impact our ability to develop and market our product candidates. Our failure to identify and correctly interpret relevant patents may negatively impact our ability to develop and market our product candidates.
One aspect of the determination of patentability of our inventions depends on the scope and content of the “prior art,” information that was or is deemed available to a person of skill in the relevant art prior to the priority date of the claimed invention. There may be prior art of which we are not aware that may affect the patentability of our patent claims or, if issued, affect the validity or enforceability of a patent claim. Further, we may not be aware of all third-party intellectual property rights potentially relating to our product candidates or their intended uses, and as a result the impact of such third-party intellectual property rights upon the patentability of our own patents and patent applications, as well as the impact of such third-party intellectual property upon our freedom to operate, is highly uncertain. Because patent applications in the United States and most other countries are confidential for typically a period of 18 months after filing, or may not be published at all, we cannot be certain that we were the first to file any patent application related to our product candidates. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Furthermore, for U.S. applications in which all claims are entitled to a priority date before March 16, 2013, an interference proceeding can be provoked by a third party or instituted by the USPTO to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. For U.S. applications containing a claim not entitled to priority before March 16, 2013, there is a greater level of uncertainty in the patent law in view of the passage of the America Invents Act, which brought into effect significant changes to the U.S. patent laws, including new procedures for challenging pending patent applications and issued patents.
| 44 |
Our patents or pending patent applications may be challenged in the courts or patent offices in the United States and abroad. For example, we may be subject to a third-party pre-issuance submission of prior art to the USPTO or become involved in PGR procedures, oppositions, derivations, reexaminations or IPR proceedings, in the United States or elsewhere, challenging our patent rights or the patent rights of others. An adverse determination in any such challenges may result in loss of exclusivity or in patent claims being narrowed, invalidated, or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. In addition, given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. Any failure to obtain or maintain patent protection with respect to our product candidates could have a material adverse effect on our business, financial condition, results of operations and prospects.
In the future, some of our intellectual property may be discovered through government-funded programs and thus may be subject to federal regulations such as “march-in” rights, certain reporting requirements and a preference for U.S.-based companies. Compliance with such regulations may limit our exclusive rights and limit our ability to contract with non-U.S. manufacturers.
Some of the intellectual property rights we may acquire or license in the future may be generated through the use of U.S. government funding and may therefore be subject to certain federal regulations. These U.S. government rights may include retained rights in the intellectual property, including a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government may have the right, under certain limited circumstances, to require us to grant exclusive, partially exclusive, or non-exclusive licenses to any of these inventions to a third party if it determines that: (i) adequate steps have not been taken to commercialize the invention; (ii) government action is necessary to meet public health or safety needs; or (iii) government action is necessary to meet requirements for public use under federal regulations (also referred to as “march-in rights”). The U.S. government may also have the right to take title to these inventions if the grant recipient fails to disclose the invention to the government or fails to file an application to register the intellectual property within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us to expend substantial resources. In addition, the U.S. government requires that any products embodying any of these inventions or produced through the use of any of these inventions be manufactured substantially in the United States. This preference for U.S. industry may be waived by the federal agency that provided the funding if the owner or assignee of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. industry may limit our ability to contract with non-U.S. product manufacturers for products covered by such intellectual property. To the extent any of our future intellectual property is also generated through the use of U.S. government funding, the provisions of the Bayh-Dole Act may similarly apply.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
| ● | others may be able to develop products that are similar to our product candidates but that are not covered by the claims of the patents that we own or license; |
| ● | we or our licensors or future collaborators might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own or license; |
| ● | we or our licensors or future collaborators might not have been the first to file patent applications covering certain of our inventions; |
| 45 |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| ● | it is possible that our licensors’ pending patent applications will not lead to issued patents; |
| ● | issued patents that we own or license may be held invalid or unenforceable, as a result of legal challenges by our competitors; |
| ● | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| ● | we may not develop additional proprietary technologies that are patentable; |
| ● | we cannot predict the scope of protection of any patent issuing based on our patent applications, including whether the patent applications that we own or in-license will result in issued patents with claims that cover our product candidates or uses thereof in the United States or in other foreign countries; |
| ● | the claims of any patent issuing based on our patent applications may not provide protection against competitors or any competitive advantages, or may be challenged by third parties; |
| ● | if enforced, a court may not hold that our patents are valid, enforceable and infringed; |
| ● | we may need to initiate litigation or administrative proceedings to enforce and/or defend our patent rights which will be costly whether we win or lose; |
| ● | we may choose not to file a patent application in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent application and obtain an issued patent covering such intellectual property; |
| ● | we may fail to adequately protect and police our trademarks and trade secrets; and |
| ● | the patents of others may have an adverse effect on our business, including if others obtain patents claiming subject matter similar to or improving that covered by our patents and patent applications. |
Should any of these events occur, they could significantly harm our business, results of operations and prospects.
Our commercial success depends significantly on our ability to operate without infringing the patents and other proprietary rights of third parties. Claims by third parties that we infringe their proprietary rights may result in liability for damages or prevent or delay our developmental and commercialization efforts.
Our commercial success depends in part on avoiding infringement of the patents and proprietary rights of third parties. However, our research, development and commercialization activities may be subject to claims that we infringe or otherwise violate patents or other intellectual property rights owned or controlled by third parties. Other entities may have or obtain patents or proprietary rights that could limit our ability to make, use, sell, offer for sale or import our product candidates and products that may be approved in the future, or impair our competitive position. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biopharmaceutical industry, including patent infringement lawsuits, oppositions, reexaminations, IPR proceedings and PGR proceedings before the USPTO and/or foreign patent offices. Numerous third-party U.S. and foreign issued patents and pending patent applications exist in the fields in which we are developing product candidates. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates.
| 46 |
As the biopharmaceutical industry expands and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement of the patent rights of third parties. We cannot provide any assurances that third-party patents do not exist which might be enforced against our current product candidates or future products, resulting in either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties. Because patent applications are maintained as confidential for a certain period of time, until the relevant application is published, we may be unaware of third-party patents that may be infringed by commercialization of any of our product candidates, and we cannot be certain that we were the first to file a patent application related to a product candidate or technology. Moreover, because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates may infringe. In addition, identification of third-party patent rights that may be relevant to our technology is difficult because patent searching is imperfect due to differences in terminology among patents, incomplete databases and the difficulty in assessing the meaning of patent claims. It is also possible that patents owned by third parties of which we are aware, but which we do not believe are relevant to our product candidates and other proprietary technologies we may develop, could be found to be infringed by our product candidate. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Any claims of patent infringement asserted by third parties would be time consuming and could:
| ● | result in costly litigation that may cause negative publicity; |
| ● | divert the time and attention of our technical personnel and management; |
| ● | cause development delays; |
| ● | prevent us from commercializing any of our product candidates until the asserted patent expires or is held finally invalid or unenforceable or not infringed in a court of law; |
| ● | require us to develop non-infringing technology, which may not be possible on a cost-effective basis; |
| ● | subject us to significant liability to third parties; or |
| ● | require us to enter into royalty or licensing agreements, which may not be available on commercially reasonable terms, or at all, or which might be non-exclusive, which could result in our competitors gaining access to the same technology. |
Although, to our knowledge, no third party has asserted a claim of patent infringement against us as of the date of this prospectus, others may hold proprietary rights that could prevent our product candidates from being marketed. Any patent-related legal action against us claiming damages and seeking to enjoin activities relating to our product candidates or processes could subject us to potential liability for damages, including treble damages if we were determined to willfully infringe, and require us to obtain a license to manufacture or develop our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of management and employee resources from our business. We cannot predict whether we would prevail in any such actions or that any license required under any of these patents would be made available on commercially acceptable terms, if at all. Moreover, even if we or our future strategic partners were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. In addition, we cannot be certain that we could redesign our product candidates or processes to avoid infringement, if necessary. Accordingly, an adverse determination in a judicial or administrative proceeding, or the failure to obtain necessary licenses, could prevent us from developing and commercializing our product candidates, which could harm our business, financial condition and operating results.
Parties making claims against us may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources or more mature and developed intellectual property portfolios, or both. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise additional funds or otherwise have a material adverse effect on our business, our ability to compete in the marketplace, results of operations, financial condition and prospects.
| 47 |
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful. Further, our in-licensed issued patents could be found invalid or unenforceable if challenged in court.
Competitors may infringe our patents or other intellectual property rights or the intellectual property rights of our licensors. To cease such infringement or unauthorized use, we and/or our licensors may be required to file infringement claims, which can be expensive and time-consuming. Further, our licensors may need to file infringement claims, and our licensors may elect not to file such claims. Our pending patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications. In addition, in a patent infringement proceeding, a court may decide that a patent we own or license is not valid, is unenforceable and/or is not infringed. If we or any of our licensors or potential future collaborators were to initiate legal proceedings against a third party to enforce a patent directed at one of our product candidates, the defendant could counterclaim that our patent is invalid and/or unenforceable in whole or in part. In patent litigation, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include an alleged failure to meet any of several statutory requirements, including lack of novelty or written description, obviousness, written description, or non-enablement. Grounds for an unenforceability assertion could include an allegation that someone connected with prosecution of the patent intentionally withheld material information from the USPTO or made a misleading statement during prosecution.
If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on such product candidate. There is also a risk that, even if the validity of such patents is upheld, the court will construe the patent’s claims narrowly or decide that we do not have the right to stop the other party from using the invention at issue on the grounds that our patent claims do not cover the invention, or decide that the other party’s use of our patented technology falls under the safe harbor to patent infringement under 35 U.S.C. §271(e)(1). In addition, if the breadth or strength of protection provided by our patents and patent applications or those of our licensors is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates. Such a loss of patent protection would have a material adverse impact on our business. Similarly, if we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the marks in question. In this case, we could ultimately be forced to cease use of such trademarks.
Even if resolved in our favor, litigation or other legal proceedings relating to our intellectual property rights may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could compromise our ability to compete in the marketplace.
Even if we establish infringement, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or other legal proceedings relating to our intellectual property rights, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation or other proceedings.
Intellectual property litigation may lead to unfavorable publicity that harms our reputation and causes the market price of our common shares to decline.
During the course of any intellectual property litigation, there could be public announcements of the initiation of the litigation as well as results of hearings, rulings on motions, and other interim proceedings in the litigation. If securities analysts or investors regard these announcements as negative, the perceived value of our existing product candidates, programs or intellectual property could be diminished. Such announcements could also harm our reputation or the market for our future product candidates, which could have a material adverse effect on our business.
| 48 |
Derivation or interference proceedings may be necessary to determine priority of inventions, and an unfavorable outcome may require us to cease using the related technology or to attempt to license rights from the prevailing party.
Derivation or interference proceedings provoked by third parties or brought by us or our licensors, or declared by the USPTO or similar proceedings in foreign patent offices may be necessary to determine the priority of inventions with respect to, or correct the inventorship of, our or our licensors’ patents or patent applications. An unfavorable outcome could result in a loss of our current patent rights and require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our or our licensors’ defense of such proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In addition, the uncertainties associated with such proceedings could have a material adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our research programs, license necessary technology from third parties or enter into development or manufacturing partnerships that would help us bring our product candidates to market.
Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
In September 2011, the Leahy-Smith America Invents Act, or Leahy-Smith Act, was signed into law. The Leahy-Smith Act could increase uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art, and provide more efficient and cost-effective avenues for competitors to challenge the validity of patents. In particular, under the Leahy-Smith Act, the United States transitioned in March 2013 to a “first inventor to file” system in which, assuming that other requirements of patentability are met, the first inventor to file a patent application will be entitled to the patent regardless of whether a third party was first to invent the claimed invention. A third party that files a patent application in the USPTO after March 2013 but before we file an application covering the same invention, could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by such third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application. Furthermore, our ability to obtain and maintain valid and enforceable patents depends on whether the differences between our technology and the prior art allow our technology to be patentable over the prior art. Since patent applications in the United States and most other countries are confidential for a period of time after filing or until issuance, we cannot be certain that we or our licensors were the first to either (i) file any patent application related to our product candidates and other proprietary technologies we may develop or (ii) invent any of the inventions claimed in our or our licensors’ patents or patent applications. Even where we have a valid and enforceable patent, we may not be able to exclude others from practicing the claimed invention where the other party can show that they used the invention in commerce before our filing date or the other party benefits from a compulsory license.
The Leahy-Smith Act also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including PGR, IPR, and derivation proceedings. An adverse determination in any such submission or proceeding could reduce the scope or enforceability of, or invalidate, our patent rights, which could adversely affect our competitive position.
Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. Thus, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our or licensors’ patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
| 49 |
Changes in U.S. patent law, or laws in other countries, could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves a high degree of technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. Changes in either the patent laws or in the interpretations of patent laws in the United States and other countries may diminish our ability to protect our inventions, obtain, maintain, and enforce our intellectual property rights, and, more generally, could affect the value of our intellectual property and may increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. We cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents. In addition, Congress or other foreign legislative bodies may pass patent reform legislation that is unfavorable to us or narrows the scope of our owned and licensed patents.
For example, the U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our or our licensors’ ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the U.S. federal courts, the USPTO, or similar authorities in foreign jurisdictions, the laws and regulations governing patents could change in unpredictable ways that would weaken our or our licensors’ ability to obtain new patents or to enforce our existing patents and patents we might obtain in the future. We cannot predict how future decisions by Congress, the federal courts or the USPTO may impact the value of our patents.
We or our licensors may be subject to claims challenging the inventorship or ownership of our or our in-licensed patents and other intellectual property.
We may also be subject to claims that former employees, collaborators, or other third parties have an ownership interest in our in-licensed patents or other intellectual property as an inventor or co-inventor. The failure to name the proper inventors on a patent application can result in the patents issuing thereon being unenforceable. Inventorship disputes may arise from conflicting views regarding the contributions of different individuals named as inventors, the effects of foreign laws where foreign nationals are involved in the development of the subject matter of the patent, conflicting obligations of third parties involved in developing our product candidates or as a result of questions regarding co-ownership of potential joint inventions. For example, we may have inventorship disputes arise from conflicting obligations of consultants or others who are involved in developing our product candidates. Alternatively, or additionally, we may enter into agreements to clarify the scope of our rights in such intellectual property. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we or our licensors fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership or a right to use. Such an outcome could have a material adverse effect on our business. Even if we or our licensors are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property. Such claims could have a material adverse effect on our business, financial condition, results of operations, and prospects.
| 50 |
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years after its first effective filing date. Various extensions may be available, but the term of a patent, and the protection it affords, is limited. Even if patents directed to our product candidates are obtained, once the patent term has expired, we may be open to competition from competitive products. Given the amount of time required for the development, testing and regulatory review of product candidates, patents directed to our product candidates might expire before or shortly after such candidates are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours. In addition, although upon issuance in the United States a patent’s life can be increased based on certain delays caused by the USPTO, this increase can be reduced or eliminated based on certain delays caused by the patent applicant during patent prosecution.
If we or our licensors do not obtain patent term extension for our product candidates, our business may be materially harmed.
Depending upon the timing, duration and specifics of FDA marketing approval, if any, of our product candidates, one or more of our U.S. patents may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Amendments. The Hatch- Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. A maximum of one patent may be extended per FDA-approved product as compensation for the patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only those claims covering such approved drug product, a method for using it or a method for manufacturing it may be extended. Patent term extension may also be available in certain foreign countries upon regulatory approval of our product candidates. However, we or our licensors may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we or our licensors are unable to obtain patent term extension or restoration or the term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our revenue could be reduced, possibly materially. Further, if this occurs, our competitors may take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case. If we do not have sufficient patent life to protect our products, our business and results of operations will be adversely affected.
We may not be able to protect our intellectual property rights throughout the world.
Although we have in-licensed pending patent applications in the United States and certain other countries, filing, prosecuting and defending patents in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries, particularly certain developing countries, do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our in-licensed inventions in all countries outside the United States or from selling or importing products made using our in-licensed inventions in and into the United States or other jurisdictions. Competitors may use our in-licensed technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we or our licensors have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our product candidates, and our or our licensors patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of many foreign countries do not favor the enforcement of patents, trade secrets, and other intellectual property protection, which could make it difficult in those jurisdictions for us to stop the infringement or misappropriation of our or our licensors’ patents or other intellectual property rights, or marketing of competing products in violation of our proprietary rights. Proceedings to enforce our or our licensors’ patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our or our licensors’ patents at risk of being invalidated, held unenforceable, or interpreted narrowly and our or our licensors’ patent applications at risk of not issuing and could provoke third parties to assert claims against us. We or our licensors may not prevail in any lawsuits that we or our licensors initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Similarly, if our trade secrets are disclosed in a foreign jurisdiction, competitors worldwide could have access to our proprietary information and we may be without satisfactory recourse. Such disclosure could have a material adverse effect on our business. Accordingly, our or our licensors’ efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
| 51 |
In addition, certain countries, including China and India, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In those countries, we and our licensors may have limited remedies if patents are infringed or if we or our licensors are compelled to grant a license to a third-party, which could materially diminish the value of those patents. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or our licensors are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired, and our business, financial condition, results of operations and prospects may be adversely affected.
Because of the expense and uncertainty of litigation in certain foreign jurisdictions, we may conclude that even if a third-party is infringing our issued patents, any patents that may be issued as a result of our pending or future patent applications or other intellectual property rights, the risk-adjusted cost of bringing and enforcing such a claim or action, which typically last for years before they are concluded, may be too high or not in the best interest of our company or our stockholders, or it may be otherwise impractical or undesirable to enforce our intellectual property against some third parties. Our competitors or other third parties may be able to sustain the costs of complex patent litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. In such cases, we may decide that the monetary cost of such litigation and the diversion of the attention of our management and scientific personnel could outweigh any benefit we receive as a result of the proceedings and that the more prudent course of action is to simply monitor the situation or initiate or seek some other non-litigious action or solution. In addition, the uncertainties associated with litigation could compromise our ability to raise the funds necessary to continue our clinical trials, continue our internal research programs, in-license needed technology or other product candidates, or enter into development partnerships that would help us bring our product candidates to market.
Obtaining and maintaining our patent protection depends on compliance with various procedural, documentary, fee payment and other requirements imposed by regulations and governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to the USPTO and various foreign patent offices at various points over the lifetime of our patents and/or applications. We have systems in place to remind us to pay these fees, and we rely on third parties to pay these fees when due. Additionally, the USPTO and various foreign patent offices require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with rules applicable to the particular jurisdiction. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If such an event were to occur, it could have a material adverse effect on our business.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to the protection afforded by other types of intellectual property, we rely on the protection of our trade secrets, including unpatented know-how, technology and other proprietary information to maintain our competitive position. Although we have taken steps to protect our trade secrets and unpatented know-how, including entering into confidentiality agreements with third parties (including, but not limited to, contractors, collaborators, and outside scientific advisors), and confidential information and inventions agreements with employees, consultants, licensors and advisors, we cannot provide any assurances that all such agreements have been duly executed, and any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. We require our employees to enter into written confidentiality agreements that assign to us any inventions, developments, creative works and useful ideas of any description that are conceived of, reduced to practice or developed in the course of their employment. In addition, we require our third-party contractors to enter into a written non-disclosure agreement that requires the third party to not disclose certain of our confidential information in any manner or for any purpose other than as necessary and/or appropriate in connection with their obligations for a defined period of time, subject to certain exclusions. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. We may need to share our proprietary information, including trade secrets, with our current and future business partners, collaborators, contractors and others located in countries at heightened risk of theft of trade secrets, including through direct intrusion by private parties or foreign actors, and those affiliated with or controlled by state actors.
| 52 |
Moreover, third parties may still obtain this information or may come upon this or similar information independently, and we would have no right to prevent them from using that technology or information to compete with us. If any of these events occurs or if we otherwise lose protection for our trade secrets, the value of this information may be greatly reduced and our competitive position would be harmed. If we or our licensors do not apply for patent protection prior to such publication or if we cannot otherwise maintain the confidentiality of our proprietary technology and other confidential information, then our ability to obtain patent protection or to protect our trade secret information may be jeopardized.
We may be subject to claims that we have wrongfully hired an employee from a competitor or that we or our employees have wrongfully used or disclosed alleged confidential information or trade secrets of their former employers.
We may be subject to claims that our employees, consultants, or independent contractors have wrongfully used or disclosed confidential information of third parties.
As is common in the biopharmaceutical industry, in addition to our employees, we engage the services of consultants to assist us in the development of our product candidates. Many of these consultants, and many of our employees, were previously employed at, or may have previously provided or may be currently providing consulting services to, other biopharmaceutical companies including our competitors or potential competitors. We may become subject to claims that we, our employees, independent contractors, or consultants inadvertently or otherwise used or disclosed trade secrets or other information proprietary to their former employers or their former or current clients. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely affect our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to our management team and other employees.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our current or future trademarks or trade names may be challenged, infringed, circumvented, or declared generic or descriptive or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in our markets of interest. During trademark registration proceedings, we may receive rejections of our applications by the USPTO or in other foreign jurisdictions. Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively and our business may be adversely affected. We may license our trademarks and trade names to third parties, such as distributors. Though these license agreements may provide guidelines for how our trademarks and trade names may be used, a breach of these agreements or misuse of our trademarks and tradenames by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names.
| 53 |
Moreover, any name we have proposed to use with product candidates in the United States may need FDA approval, regardless of whether we have registered it, or applied to register it, as a trademark. Similar requirements exist in Europe. The FDA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA (or an equivalent administrative body in a foreign jurisdiction) objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA. Furthermore, in many countries, owning and maintaining a trademark registration may not provide an adequate defense against a subsequent infringement claim asserted by the owner of a senior trademark. At times, competitors or other third parties may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. If we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the marks in question. In this case, we could ultimately be forced to cease use of such trademarks.
Risks Related to Employee Matters and Managing our Growth
If we are unable to establish sales or marketing capabilities or enter into agreements with third parties to sell or market our product candidates, we may not be able to successfully sell or market our product candidates that obtain regulatory approval.
We currently do not have and have never had a marketing or sales team. In order to commercialize any product candidates, if approved, we must build marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services for each of the territories in which we may have approval to sell or market our product candidates. We may not be successful in accomplishing these required tasks.
Establishing an internal sales or marketing team with technical expertise and supporting distribution capabilities to commercialize our product candidates will be expensive and time-consuming, and will require significant attention of our executive officers to manage. Any failure or delay in the development of our internal sales, marketing and distribution capabilities could adversely impact the commercialization of any of our product candidates that we obtain approval to market, if we do not have arrangements in place with third parties to provide such services on our behalf. Alternatively, if we choose to collaborate, either globally or on a territory-by-territory basis, with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems, we will be required to negotiate and enter into arrangements with such third parties relating to the proposed collaboration. If we are unable to enter into such arrangements when needed, on acceptable terms, or at all, we may not be able to successfully commercialize any of our product candidates that receive regulatory approval, or any such commercialization may experience delays or limitations. If we are unable to successfully commercialize our approved product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
Our success is highly dependent on our ability to attract and retain highly skilled executive officers and employees.
To succeed, we must recruit, retain, manage and motivate qualified clinical, scientific, technical and management personnel, and we face significant competition for experienced personnel. We are highly dependent on the principal members of our management and scientific and medical staff. If we do not succeed in attracting and retaining qualified personnel, particularly at the management level, it could adversely affect our ability to execute our business plan and harm our operating results. In particular, the loss of one or more of our executive officers could be detrimental to us if we cannot recruit suitable replacements in a timely manner. The competition for qualified personnel in the biotechnology field is intense and as a result, we may be unable to continue to attract and retain qualified personnel necessary for the future success of our business. We could in the future have difficulty attracting experienced personnel to our company and may be required to expend significant financial resources in our employee recruitment and retention efforts.
Many of the other biotechnology companies that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. They also may provide more diverse opportunities and better prospects for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what we have to offer. If we are unable to continue to attract and retain high-quality personnel, the rate and success at which we can discover, develop and commercialize our product candidates will be limited and the potential for successfully growing our business will be harmed.
| 54 |
In order to successfully implement our plans and strategies, we will need to grow the size of our organization, and we may experience difficulties in managing this growth.
As of September 30, 2023, we had eight full-time employees. In order to successfully implement our development and commercialization plans and strategies, we expect to need additional managerial, operational, sales, marketing, financial and other personnel. Future growth would impose significant added responsibilities on members of management, including:
| ● | identifying, recruiting, integrating, maintaining and motivating additional employees; |
| ● | managing our internal development efforts effectively, including the clinical, FDA, EMA and other comparable foreign regulatory agencies’ review process of our product candidates and any other product candidate we develop, while complying with any contractual obligations to contractors and other third parties we may have; and |
| ● | improving our operational, financial and management controls, reporting systems and procedures. |
Our future financial performance and our ability to successfully develop and, if approved, commercialize any of our current product candidates and any other product candidate we may develop will depend, in part, on our ability to effectively manage any future growth, and our management may also have to divert a disproportionate amount of its attention away from day-to-day activities in order to devote a substantial amount of time to managing these growth activities.
We currently rely, and for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors and consultants to provide certain services, including key aspects of clinical development and manufacturing. We cannot assure you that the services of independent organizations, advisors and consultants will continue to be available to us on a timely basis when needed, or that we can find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by third party service providers is compromised for any reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain marketing approval of any current or future product candidates or otherwise advance our business. We cannot assure you that we will be able to manage our existing third party service providers or find other competent outside contractors and consultants on economically reasonable terms, or at all.
If
we are not able to effectively expand our organization by hiring new employees and/or engaging additional third party service providers,
we may not be able to successfully implement the tasks necessary to further develop and commercialize our product candidates and any
future product candidates and, accordingly, may not achieve our research, development and commercialization goals.
Risks Related to This Offering and Ownership of Our Common Stock
The direct listing process differs from an initial public offering underwritten on a firm-commitment basis.
This is not an underwritten initial public offering of common stock. This listing of our common stock on Nasdaq differs from an underwritten initial public offering in several significant ways, which include, but are not limited to, the following:
| ● | There are no underwriters engaged on a firm-commitment basis. Consequently, prior to the opening of trading on Nasdaq, there will be no traditional book building process and no price at which underwriters initially sold shares to the public to help inform efficient and sufficient price discovery with respect to the opening trades on Nasdaq. Therefore, buy and sell orders submitted prior to and at the opening of trading of our common stock on Nasdaq will not have the benefit of being informed by a published price range or a price at which the underwriters initially sold shares to the public, as would be the case in an initial public offering underwritten on a firm-commitment basis. Moreover, there will be no underwriters engaged on a firm-commitment underwritten basis assuming risk in connection with the initial resale of shares of our common stock. In an initial public offering underwritten on a firm-commitment basis, the underwriters may engage in “covered” short sales in an amount of shares representing the underwriters’ option to purchase additional shares. To close a covered short position, the underwriters purchase shares in the open market or exercise the underwriters’ option to purchase additional shares. In determining the source of shares to close the covered short position, the underwriters typically consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the underwriters’ option to purchase additional shares. Purchases in the open market to cover short positions, as well as other purchases underwriters may undertake for their own accounts, may have the effect of preventing a decline in the market price of shares. Given that there will be no underwriters’ option to purchase additional shares and no underwriters engaging in stabilizing transactions, there could be greater volatility in the public price of our common stock during the period immediately following the listing. See also “— Our shares of common stock have no prior public market. An active trading market may not develop or continue to be liquid and the market price of our shares of common stock may be volatile.” |
| 55 |
| ● | There is not a fixed number of shares of common stock available for sale. Therefore, there can be no assurance that any Registered Stockholders or other existing stockholders will sell any or all of their common stock and there may initially be a lack of supply of, or demand for, our common stock on Nasdaq. Alternatively, we may have a large number of Registered Stockholders or other existing stockholders who choose to sell their common stock in the near term resulting in an oversupply of our common stock, which could adversely impact the public price of our common stock once listed on Nasdaq and thereafter. |
| ● | None of our Registered Stockholders or other existing stockholders have entered into contractual lock-up agreements or other contractual restrictions on transfer. In a firm-commitment underwritten initial public offering, it is customary for an issuer’s officers, directors, and most of its other stockholders to enter into a 180-day contractual lock-up arrangement with the underwriters to help promote orderly trading immediately after such initial public offering. Consequently, any of our stockholders, including our directors and officers who own our common stock and other significant stockholders, may sell any or all of their common stock at any time (subject to any restrictions under applicable law), including immediately upon listing. If such sales were to occur in a significant volume in a short period of time following our listing, it may result in an oversupply of our common stock in the market, which could adversely impact the public price of our common stock. |
| ● | We will not conduct a traditional “roadshow” with underwriters prior to the opening of trading on Nasdaq. Instead, we intend to host an investor day, as well as engage in certain other investor education meetings. In advance of the investor day, we will announce the date for such day over financial news outlets in a manner consistent with typical corporate outreach to investors. We will prepare an electronic presentation for this investor day, which will have content similar to a traditional roadshow presentation, and make one version of the presentation publicly available, without restriction, on a website. There can be no guarantees that the investor day and other investor education meetings will have the same impact on investor education as a traditional “roadshow” conducted in connection with a firm-commitment underwritten initial public offering. As a result, there may not be efficient price discovery with respect to our common stock or sufficient demand among investors immediately after our listing, which could result in a more volatile public price of our common stock. |
Such differences from a firm-commitment underwritten initial public offering could result in a volatile trading price for our common stock and uncertain trading volume, which may adversely affect your ability to sell any common stock that you may purchase.
Our common stock currently has no public market. An active trading market may not develop or continue to be liquid and the market price of shares of our common stock may be volatile.
We expect our common stock to be listed and traded on Nasdaq. Prior to the listing on Nasdaq, there has not been a public market for any of our securities, and an active market for our common stock may not develop or be sustained after the listing, which could depress the market price of shares of our common stock and could affect the ability of our stockholders to sell our common stock. In the absence of an active public trading market, investors may not be able to liquidate their investments in our common stock. An inactive market may also impair our ability to raise capital by selling shares of our common stock, our ability to motivate our employees through equity incentive awards and our ability to acquire other companies, products or technologies by using shares of our common stock as consideration.
| 56 |
In addition, we cannot predict the prices at which our common stock may trade on Nasdaq following the listing of our common stock, and the market price of our common stock may fluctuate significantly in response to various factors, some of which are beyond our control. In particular, as this listing is taking place through a novel process that is not a firm-commitment underwritten initial public offering, there will be no traditional book building process and no price at which traditional underwriters initially sold shares to the public to help inform efficient price discovery with respect to the opening trades on Nasdaq. On the day that our shares of common stock are initially listed on Nasdaq, Nasdaq will begin accepting, but not executing, pre-opening buy and sell orders and will begin to continuously generate the indicative Current Reference Price on the basis of such accepted orders. The Current Reference Price is calculated each second and, during a 10-minute “Display Only” period, is disseminated, along with other indicative imbalance information, to market participants by Nasdaq on its NOII and BookViewer tools. Following the “Display Only” period, a “Pre-Launch” period begins, during which the Advisor, in its capacity as our financial advisor, must notify Nasdaq that our shares are “ready to trade.” Once the Advisor has notified Nasdaq that our shares of common stock are ready to trade, Nasdaq will confirm the Current Reference Price for our shares of common stock, in accordance with Nasdaq rules. If the Advisor then approves proceeding at the Current Reference Price, the applicable orders that have been entered will be executed at such price and regular trading of shares of our common stock on Nasdaq will commence, subject to Nasdaq conducting validation checks in accordance with Nasdaq rules. The Advisor will determine when our shares of common stock are ready to trade and approve proceeding at the Current Reference Price primarily based on considerations of volume, timing and price. In particular, the Advisor will determine, based primarily on pre-opening buy and sell orders, when a reasonable amount of volume will cross on the opening trade such that sufficient price discovery has been made to open trading at the Current Reference Price. If the Advisor does not approve proceeding at the Current Reference Price (for example, due to the absence of adequate preopening buy and sell interest), the Advisor will request that Nasdaq delay the open until such a time that sufficient price discovery has been made to ensure a reasonable amount of volume crosses on the opening trade. For more information, see “Plan of Distribution.”
Additionally, prior to the opening trade, there will not be a price at which underwriters initially sold shares of common stock to the public as there would be in a firm-commitment underwritten initial public offering. The absence of a predetermined initial public offering price could impact the range of buy and sell orders collected by Nasdaq from various broker-dealers. Consequently, upon listing on Nasdaq, the public price of our common stock may be more volatile than in a firm-commitment underwritten initial public offering and could decline significantly and rapidly.
Furthermore, because of our novel listing process on Nasdaq, Nasdaq’s rules for ensuring compliance with its initial listing standards, such as those requiring a valuation or other compelling evidence of value, are untested. In the absence of a prior active public trading market for our common stock, if the price of our common stock or our market capitalization falls below those required by Nasdaq’s eligibility standards, we may not be able to satisfy the ongoing listing criteria and may be required to delist.
In addition, because of our novel listing process, individual investors, retail or otherwise, may have greater influence in setting the opening public price and subsequent public prices of our common stock on Nasdaq and may participate more in our initial trading than is typical for a firm-commitment underwritten initial public offering. These factors could result in a public price of our common stock that is higher than other investors (such as institutional investors) are willing to pay, which could cause volatility in the trading price of our common stock and an unsustainable trading price if the price of our common stock significantly rises upon listing and institutional investors believe our common stock is worth less than retail investors, in which case the price of our common stock may decline over time. Further, if the public price of our common stock is above the level that investors determine is reasonable for our common stock, some investors may attempt to short our common stock after trading begins, which would create additional downward pressure on the public price of our common stock. To the extent that there is a lack of consumer awareness among retail investors, such a lack of consumer awareness could reduce the value of our common stock and cause volatility in the trading price of our common stock.
| 57 |
The public price of our common stock following the listing also could be subject to wide fluctuations in response to the risk factors described in this prospectus and others beyond our control, including:
| ● | changes in the industries in which we operate; |
| ● | variations in our operating performance and the performance of our competitors in general; |
| ● | actual or anticipated fluctuations in our quarterly or annual operating results; |
| ● | publication of research reports by securities analysts about us or our competitors or our industry; |
| ● | the public’s reaction to our press releases, our other public announcements and our filings with the SEC; |
| ● | our failure or the failure of our competitors to meet analysts’ projections or guidance that we or our competitors may give to the market; |
| ● | additions and departures of key personnel; |
| ● | changes in laws and regulations affecting our business; |
| ● | commencement of, or involvement in, litigation involving us; |
| ● | changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
| ● | the volume of shares of our common stock available for public sale; and |
| ● | general economic and political conditions such as recessions, interest rates, fuel prices, foreign currency fluctuations, international tariffs, social, political and economic risks and acts of war or terrorism. |
In addition, securities exchanges have experienced price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies. Stock prices of many companies have fluctuated in a manner often unrelated to the operating performance of those companies. These fluctuations may be even more pronounced in the trading market for our common stock shortly following the listing of our common stock on Nasdaq as a result of the supply and demand forces described above. In the past, stockholders have instituted securities class action litigation following periods of market volatility. If we were to become involved in securities litigation, it could subject us to substantial costs, divert resources and the attention of management from our business and harm our business, results of operations and financial condition.
Future sales of common stock by our Registered Stockholders and other existing stockholders could cause our share price to decline.
We currently expect our common stock to be listed and traded on Nasdaq. Prior to listing on Nasdaq, there has been no public market for our common stock and there has not been a sustained history of trading in our common stock in “over-the-counter” markets. While our common stock may be sold after our listing on Nasdaq by the Registered Stockholders pursuant to this prospectus or by our other existing stockholders in accordance with Rule 144 under the Securities Act, unlike a firm-commitment underwritten initial public offering, there can be no assurance that any Registered Stockholders or other existing stockholders will sell any of their shares of common stock and there may initially be a lack of supply of, or demand for, common stock on Nasdaq. As described herein, certain shares of our common stock outstanding as of the date hereof will be registered under this registration statement. There can be no assurance that the Registered Stockholders and other existing stockholders will not sell all of their shares of common stock, resulting in an oversupply of our common stock on Nasdaq. In the case of a lack of supply of our common stock, the trading price of our common stock may rise to an unsustainable level. Further, institutional investors may be discouraged from purchasing our common stock if they are unable to purchase a block of our common stock in the open market due to a potential unwillingness of our existing stockholders to sell a sufficient amount of common stock at the price offered by such institutional investors and the greater influence individual investors have in setting the trading price. If institutional investors are unable to purchase our common stock, the market for our common stock may be more volatile without the influence of long-term institutional investors holding significant amounts of our common stock. In the case of a lack of market demand for our common stock, the trading price of our common stock could decline significantly and rapidly after our listing. Therefore, an active, liquid and orderly trading market for our common stock may not initially develop or be sustained, which could significantly depress the public price of our common stock and/or result in significant volatility, which could affect your ability to sell your shares of common stock.
| 58 |
Upon the Direct Listing, we will have 2,500 shares of Series C Preferred Stock with super voting rights.
Our capital stock as of the date hereof consists of voting common stock (which we sometimes refer to herein as “common stock”), non-voting common stock, Series A Preferred Stock, Series B Preferred Stock and Series B-1 Preferred Stock. Our board of directors and stockholders have each approved the creation and issuance of an aggregate of 2,500 (after giving effect to the Reverse Stock Split) Series C Preferred Stock, all of which Series C Preferred Stock will be issued to Pete O’Heeron, our founder and Chief Executive Officer, such that, prior to the Direct Listing, our capital stock will consist of common stock, non-voting common stock, Series B Preferred, Series B-1 Preferred Stock and Series C Preferred Stock.
In connection with the Direct Listing, (i) all of our then outstanding Series A Preferred Stock, all of which are held by FibroGenesis, will be automatically canceled without the payment of additional consideration by or to the holder thereof, (ii) all of our then outstanding non-voting common stock will automatically convert, without the payment of additional consideration by or to the holders thereof, into common stock, on a one-for-one basis, (iii) all of our then outstanding Series B Preferred Stock and all of our then outstanding Series B-1 Preferred Stock will automatically convert, without the payment of additional consideration by or to the holders thereof, into common stock, on a one-for-one basis and (iv) all of our then outstanding Series C Preferred Stock will remain Series C Preferred Stock, such that, immediately after the Direct Listing, our issued and outstanding capital stock will consist of common stock and Series C Preferred Stock.
The Series C Preferred Stock (i) have no dividend rights, (ii) convert into common stock upon any transfer from the initial holder, (iii) have a liquidation preference of $18.00 per share (subject to appropriate adjustment in the event of any stock split, combination, or other similar recapitalization) upon our liquidation, dissolution or winding up and (iv) upon the Direct Listing, will be entitled to 13,000 votes for each share of Series C Preferred Stock (prior to the Direct Listing, the Series C Preferred will have no right to vote on any matter except as required by Delaware law, and in such required case, will have one vote per share of Series C Preferred Stock).
The Series C Preferred Stock will be subject to an irrevocable proxy issued by Pete O’Heeron, the holder of all of the Series C Preferred Stock, in favor and for the benefit of, our board of directors, granting our board of directors the irrevocable proxy, for as long as the Series C Preferred Stock remain outstanding, to vote all of the Series C Preferred Stock on all matters on which the Series C Preferred Stock are entitled to vote, in any manner that our board of directors may determine in its sole and absolute discretion; provided, however, that such irrevocable proxy shall not, without the written consent of Pete O’Heeron, permit our board of directors to vote the Series Preferred Stock with respect to any proposal to amend, delete or waive any rights of Pete O’Heeron with respect to the Series C Preferred Stock as set forth in our amended and restated certificate of incorporation. In light of the superior voting rights associated with the Series C Preferred Stock, the irrevocable proxy is intended to ensure that such superior voting rights are utilized in our best interest and to avoid or mitigate conflicts that may arise in the future for Pete O’Heeron as an individual stockholder employee.
See “Description of Capital Stock—Series C Preferred Stock” for additional information regarding our Series C Preferred Stock.
In addition to the dilutive effect on the voting power and value of our common stock, the foregoing structure of our capital stock may render our common stock ineligible for inclusion in certain securities market indices, and thus adversely affect the price and liquidity of, and public sentiment regarding, our common stock or other securities. The existence of, and voting rights associated with, our Series C Preferred Stock, either alone or in conjunction with certain of the other provisions of our amended and restated certificate of incorporation, such as the requirement to have a staggered board, could also have the effect of delaying, deterring or preventing a change in our control or make the removal of our management more difficult.
We will be a “controlled company” within the meaning of the Nasdaq Stock Market Rules upon the Direct Listing because our insiders will beneficially own more than 50% of the voting power of our outstanding voting securities.
Upon completion of this offering, our founder and Chief Executive Officer, Pete O’Heeron, will collectively beneficially own approximately 59% of the voting power of our outstanding voting securities and we will be a “controlled company” within the meaning of the listing rules of The Nasdaq Stock Market LLC. We may rely on certain exemptions from corporate governance rules, including an exemption from the rule that a majority of our board of directors must be independent directors. Although we currently do not intend to rely on the “controlled company” exemption under the Nasdaq listing rules, we could elect to rely on this exemption in the future. In the event that we elected to rely on the “controlled company” exemption, a majority of the members of our board of directors might not be independent directors, and our nominating and corporate governance and compensation committees might not consist entirely of independent directors. Our status as a controlled company could cause our shares of common stock to be less attractive to certain investors or otherwise harm our trading price. As a result, you would not have the same protection afforded to shareholders of companies that are subject to these corporate governance requirements.
You may be diluted by future issuances of preferred stock or additional common stock in connection with our incentive plans, acquisitions or otherwise; future sales of such shares in the public market, or the expectations that such sales may occur, could lower our stock price.
Prior to the effectiveness of the registration statement of which this prospectus forms a part, we will adopt an amended and restated certificate of incorporation which will authorize us to issue shares of common stock and options, rights, warrants and appreciation rights relating to our common stock for the consideration and on the terms and conditions established by our board of directors in its sole discretion. We could issue a significant number of shares of common stock in the future in connection with investments or acquisitions. Any of these issuances could dilute our existing stockholders, and such dilution could be significant. Moreover, such dilution could have a material adverse effect on the market price for the shares of our common stock.
The future issuance of shares of preferred stock with voting rights may adversely affect the voting power of the holders of shares of our common stock, either by diluting the voting power of our common stock if the preferred stock votes together with the common stock as a single class, or by giving the holders of any such preferred stock the right to block an action on which they have a separate class vote, even if the action were approved by the holders of our shares of our common stock.
The future issuance of shares of preferred stock with dividend or conversion rights, liquidation preferences or other economic terms favorable to the holders of preferred stock could adversely affect the market price for our common stock by making an investment in the common stock less attractive. For example, investors in the common stock may not wish to purchase common stock at a price above the conversion price of a series of convertible preferred stock because the holders of the preferred stock would effectively be entitled to purchase common stock at the lower conversion price, causing economic dilution to the holders of common stock.
Because
we have no current plans to pay cash dividends on our common stock, you may not receive any return on investment unless you sell your
common stock for a price greater than that which you paid for it.
We currently intend to retain all available funds and any future earnings to fund the development, commercialization and growth of our business, and therefore we do not anticipate declaring or paying any cash dividends on our common stock in the foreseeable future. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements, general business conditions and other factors that our board of directors may deem relevant. Our future ability to pay cash dividends on our common stock may also be limited by the terms of any future debt securities or credit facility. As a result, capital appreciation, if any, of the common stock you purchase in this offering will be your sole source of gain for the foreseeable future.
We are an emerging growth company and a smaller reporting company, and the reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including (i) not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, (ii) having the option of delaying the adoption of certain new or revised financial accounting standards, (iii) reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements and (iv) exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We may take advantage of these exemptions until such time that we are no longer an emerging growth company. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold stock. Further, pursuant to Section 107 of the JOBS Act, we have elected to take advantage of the extended transition period for complying with new or revised accounting standards until those standards would otherwise apply to private companies. As a result, our operating results and financial statements may not be comparable to the operating results and financial statements of other companies who have adopted the new or revised accounting standards.
| 59 |
We will remain an emerging growth company until the earliest of (i) December 31, 2028, (ii) the last day of the fiscal year in which we have total annual gross revenue of at least $1.235 billion, (iii) the last day of the fiscal year in which we are deemed to be a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our common stock held by non-affiliates was $700.0 million or more as of the last business day of the second fiscal quarter of such year or (iv) the date on which we have issued more than $1.0 billion in non-convertible debt securities during the prior three-year period.
We are also a “smaller reporting company” as defined in the Exchange Act. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies until the fiscal year following the determination that our voting and non-voting common stock held by non-affiliates is $250 million or more measured on the last business day of our second fiscal quarter, or our annual revenues are less than $100 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is $700 million or more measured on the last business day of our second fiscal quarter.
It is possible that some investors will find our common stock less attractive as a result of the foregoing, which may result in a less active trading market for our common stock and higher volatility in our stock price.
Our management and principal stockholders own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
As of , 2023, our executive officers, directors and five percent or greater stockholders and their respective affiliates, beneficially own, in the aggregate, approximately % of our outstanding common stock on an as converted basis. To the extent that the same group continue to own a significant percentage of our common stock following this offering, these stockholders, if they act together, will be able to control the management and affairs of our company and most matters requiring stockholder approval, including the election of directors, amendments of our organizational documents and approval of any merger, sale of substantially all our assets or other significant corporate transactions. This concentration of ownership may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you or other stockholders may feel are in your or their best interest as one of our stockholders.
Provisions of our amended and restated certificate of incorporation and bylaws, in each case, which will become effective in connection with the effectiveness of the registration statement, of which this prospectus forms a part, may delay or prevent a take-over that may not be in the best interests of our stockholders.
Provisions of our amended and restated certificate of incorporation and bylaws, in each case, which will become effective in connection with the effectiveness of the registration statement, of which this prospectus forms a part, may be deemed to have anti-takeover effects, which include, among others, (i) the existence of our Series C Preferred Stock entitled to 13,000 votes per share of Series C Preferred Stock, as more particularly described elsewhere in this prospectus, (ii) a classified board of directors serving staggered three-year terms, (iii) who can fill vacancies of our board of directors, (iv) supermajority voting thresholds for the removal of members of our board, and (v) when and by whom special meetings of our stockholders may be called, and may delay, defer or prevent a takeover attempt.
In addition, our amended and restated certificate of incorporation will authorize the issuance of shares of preferred stock which will have such rights and preferences determined from time to time by our board of directors. Following the adoption of the amended and restated certificate of incorporation, our board of directors may, without stockholder approval (except as may be required under Nasdaq rules), issue additional preferred shares with dividends, liquidation, conversion, voting or other rights that could adversely affect the voting power or other rights of the holders of our common stock. Further, our amended and restated certificate of incorporation will authorize the issuance of “blank check” preferred stock that our board of directors could use to implement a stockholder rights plan (also known as a “poison pill”).
| 60 |
Our amended and restated certificate of incorporation, in each case, which will become effective in connection with the effectiveness of the registration statement, of which this prospectus forms a part, will provide for an exclusive forum in the Court of Chancery of the State of Delaware for certain disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation, in each case, which will become effective in connection with the effectiveness of the registration statement, of which this prospectus forms a part, will provide that, unless we consent in writing to the selection of an alternative forum, (i) the Court of Chancery of the State of Delaware (or, if the Court of Chancery does not have jurisdiction, the federal district court for the District of Delaware) shall, to the fullest extent permitted by law, be the sole and exclusive forum for (a) any derivative action or proceeding brought on our behalf, (b) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees to us or our stockholders, (c) any action arising pursuant to any provision of the General Corporation Law of the State of Delaware, or the DGCL, our certificate of incorporation or our bylaws or (d) any action asserting a claim governed by the internal affairs doctrine and (ii) to the fullest extent permitted by law, the federal district courts of the United States of America shall be the exclusive forum for the resolution of any complaint asserting a cause or causes of action arising under the Securities Act, including all causes of action asserted against any defendant to such complaint. Pursuant to our planned amended and restated certificate of incorporation, any person or entity purchasing or otherwise acquiring or holding any interest in shares of our common stock will be deemed to have had notice of and consented to the forum selection clause in our planned amended and restated certificate of incorporation described in this paragraph.
The foregoing provision would not preclude stockholders that assert claims under the Exchange Act from bringing such claims in federal court, to the extent that the Exchange Act confers exclusive federal jurisdiction over such claims, subject to applicable law.
We believe our choice of forum provision may benefit us by providing increased consistency in the application of Delaware law by chancellors and judges particularly experienced in resolving corporate disputes, efficient administration of cases on a more expedited schedule relative to other forums and protection against the burdens of multi-forum litigation. However, our choice of forum provision may impose additional litigation costs on stockholders in pursuing claims and may limit a stockholder’s ability to bring a claim in a judicial forum that it believes to be favorable for disputes with us or any of our directors, officers or other employees, which may discourage lawsuits with respect to such claims. In addition, while the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring a claim in a venue other than those designated in the choice of forum provision, and there can be no assurance that such provision will be enforced by a court in those other jurisdictions. If a court were to find the choice of forum provision in our certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect our business and financial condition.
General Risks
Reports published by analysts, including projections in those reports that differ from our actual results, could adversely affect the price and trading volume of our common stock.
Securities research analysts may establish and publish their own periodic projections for our Company. These projections may vary widely and may not accurately predict the results we actually achieve. The price of our common stock may decline if our actual results do not match the projections of these securities research analysts. Similarly, if one or more of the analysts who write reports on us downgrades our stock or publishes inaccurate or unfavorable research about our business, our stock price could decline. If one or more of these analysts ceases coverage of us or fails to publish reports on us regularly, our stock price or trading volume could decline.
Our internal computer systems, or those of any of our CROs, manufacturers, other contractors, consultants, collaborators or potential future collaborators, may fail or suffer security or data privacy breaches or other unauthorized or improper access to, use of, or destruction of our proprietary or confidential data, employee data or personal data, which could result in additional costs, loss of revenue, significant liabilities, harm to our brand and material disruption of our operations.
Despite the implementation of security measures, our internal computer systems and those of our current and any future CROs and other contractors, consultants, collaborators and third-party service providers, are vulnerable to damage from computer viruses, cybersecurity threats, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failure. Because the techniques used to obtain unauthorized access to, or to sabotage, systems change frequently and often are not recognized until launched against a target, we may be unable to anticipate these techniques or implement adequate preventative measures. We may also experience security breaches that may remain undetected for an extended period. If such an event were to occur and cause interruptions in our operations or result in the unauthorized acquisition of or access to personally identifiable information or individually identifiable health information (violating certain privacy laws such as HIPAA and GDPR), it could result in a material disruption of our drug discovery and development programs and our business operations, whether due to a loss of our trade secrets or other similar disruptions. Some of the federal, state and foreign government requirements include obligations of companies to notify individuals of security breaches involving particular personally identifiable information, which could result from breaches experienced by us or by our vendors, contractors or organizations with which we have formed strategic relationships. Notifications and follow-up actions related to a security breach could impact our reputation, cause us to incur significant costs, including legal expenses and remediation costs. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the lost data. We also rely on third parties for certain portions of our manufacturing process, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data, or inappropriate disclosure of confidential or proprietary information, we could be exposed to litigation and governmental investigations, the further development and commercialization of our product candidates could be delayed, and we could be subject to significant fines or penalties for any noncompliance with certain state, federal and/or international privacy and security laws.
Our insurance policies may not be adequate to compensate us for the potential losses arising from any such disruption, failure or security breach. In addition, such insurance may not be available to us in the future on economically reasonable terms, or at all. Further, our insurance may not cover all claims made against us and could have high deductibles in any event, and defending a suit, regardless of its merit, could be costly and divert management attention.
Our operations are vulnerable to interruption by fire, severe weather conditions, power loss, telecommunications failure, terrorist activity and other events beyond our control, which could harm our business.
Our facility is located in a region which experiences severe weather from time to time. We have not undertaken a systematic analysis of the potential consequences to our business and financial results from a major tornado, flood, fire, earthquake, power loss, terrorist activity or other disasters and do not have a recovery plan for such disasters. In addition, we do not carry sufficient insurance to compensate us for actual losses from interruption of our business that may occur, and any losses or damages incurred by us could harm our business. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
| 61 |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements that can involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this prospectus, including statements regarding our future results of operations and financial position, business strategy, prospective products, product approvals, research and development costs, future revenue, timing and likelihood of success, plans and objectives of management for future operations, future results of anticipated products and prospects, plans and objectives of management are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” or “would” or the negative of these terms or other similar expressions, although not all forward-looking statements contain these words. Forward-looking statements contained in this prospectus include, but are not limited to, statements about:
| ● | the timing, progress and results of preclinical studies and clinical trials for our current and future product candidates, including statements regarding the timing of initiation and completion of studies or trials and related preparatory work, the period during which the results of the trials will become available and our research and development programs; |
| ● | the timing, scope or likelihood of regulatory submissions, filings, and approvals, including final regulatory approval of our product candidates; |
| ● | our ability to develop and advance product candidates into, and successfully complete, clinical trials; |
| ● | our expectations regarding the size of the patient populations for our product candidates, if approved for commercial use; |
| ● | the implementation of our business model and our strategic plans for our business, product candidates and technology; |
| ● | our commercialization, marketing and manufacturing capabilities and strategy; |
| ● | the pricing and reimbursement of our product candidates, if approved; |
| ● | the rate and degree of market acceptance and clinical utility of our product candidates, in particular, and cell therapy, in general; |
| ● | our ability to establish or maintain collaborations or strategic relationships or obtain additional funding; |
| ● | our competitive position; |
| ● | the scope of protection we and/or our licensors are able to establish and maintain for intellectual property rights covering our product candidates; |
| ● | developments and projections relating to our competitors and our industry; |
| ● | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; |
| ● | the period over which we estimate our existing cash and cash equivalents will be sufficient to fund our future operating expenses and capital expenditure requirements; and |
| ● | the impact of laws and regulations. |
We have based these forward-looking statements largely on our current expectations and projections about our business, the industry in which we operate and financial trends that we believe may affect our business, financial condition, results of operations and prospects, and these forward-looking statements are not guarantees of future performance or development. These forward-looking statements speak only as of the date of this prospectus and are subject to a number of risks, uncertainties and assumptions described in the section titled “Risk Factors” and elsewhere in this prospectus. Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein until after we distribute this prospectus, whether as a result of any new information, future events or otherwise.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this prospectus, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and you are cautioned not to unduly rely upon these statements.
| 62 |
This prospectus includes estimates regarding market and industry data. Unless otherwise indicated, information concerning our industry and the markets in which we operate, including our general expectations, market position, market opportunity, and market size, are based on our management’s knowledge and experience in the markets in which we operate, together with currently available information obtained from various sources, including publicly available information, industry reports and publications, surveys, our customers, trade and business organizations, and other contacts in the markets in which we operate. Certain information is based on management estimates, which have been derived from third-party sources, as well as data from our internal research.
In presenting this information, we have made certain assumptions that we believe to be reasonable based on such data and other similar sources and on our knowledge of, and our experience to date in, the markets in which we operate. While we believe the estimated market and industry data included in this prospectus is generally reliable, such information is inherently uncertain and imprecise. Market and industry data is subject to change and may be limited by the availability of raw data, the voluntary nature of the data gathering process, and other limitations inherent in any statistical survey of such data. In addition, projections, assumptions, and estimates of the future performance of the markets in which we operate are necessarily subject to uncertainty and risk due to a variety of factors, including those described in “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements.” These and other factors could cause results to differ materially from those expressed in the estimates made by third parties and by us. Accordingly, you are cautioned not to place undue reliance on such market and industry data or any other such estimates.
The source of certain statistical data, estimates, and forecasts contained in this prospectus are the following independent industry publications or reports:
| ● | “Degenerative Disc Disease Therapeutics Global Market Analysis, Insights and Forecast, 2022-2029” Fortune Business Insights; | |
| ● | “Global Regenerative Medicine Market 2022-2029” Fortune Business Insights; | |
| ● | “Global Multiple Sclerosis Drugs Market 2022-2029” Fortune Business Insights; and | |
| ● | “Global Wound Care Market 2022-2029” Fortune Business Insights. |
The content of the above sources, except to the extent specifically set forth in this prospectus, does not constitute a portion of this prospectus and is not incorporated herein.
| 63 |
TRADEMARKS, SERVICE MARKS AND TRADENAMES
We own or otherwise have rights to the trademarks, including those mentioned in this prospectus, used in conjunction with the operation of our business. This prospectus includes our own trademarks, which are protected under applicable intellectual property laws, as well as trademarks, service marks and tradenames of other entities, which are the property of their respective owners. Solely for convenience, trademarks, trade names and service marks referred to in this prospectus may appear without the ®, TM or SM symbols, but such references are not intended to indicate, in any way, that the applicable licensor will not assert, to the fullest extent under applicable law, its rights to these trademarks, service marks and tradenames. We do not intend our use or display of other entities’ trademarks, service marks or tradenames to imply a relationship with, or endorsement or sponsorship of us by, any other entities.
| 64 |
The Registered Stockholders may, or may not, elect to sell shares of our common stock covered by this prospectus. To the extent any Registered Stockholder chooses to sell shares of our common stock covered by this prospectus, we will not receive any proceeds from any such sales of our common stock. See “Principal and Registered Stockholders.”
We have never declared or paid cash dividends on our common stock. We currently intend to retain all available funds and any future earnings to fund the development, commercialization and growth of our business, and therefore we do not anticipate declaring or paying any cash dividends on our common stock in the foreseeable future. Any future determination as to the declaration and payment of dividends, if any, will be at the discretion of our board of directors. Any such determination will also depend upon our business prospects, operating results, financial condition, capital requirements, general business conditions and other factors that our board of directors may deem relevant. Our future ability to pay cash dividends on our common stock may also be limited by the terms of any future debt securities or credit facility.
The following table sets forth our cash and cash equivalents and capitalization as of September 30, 2023, as follows.
| ● | on an actual basis (after giving effect to the Reverse Stock Split and the Capital Stock Adjustments); | |
| ● | on a pro forma basis to give effect to (i) the automatic elimination of our Series A Preferred Stock for no consideration in connection with the Direct Listing, (ii) the automatic conversion, without any consideration, of all outstanding shares of our convertible preferred stock (being our Series B Preferred Stock and our Series B-1 Preferred Stock) and all outstanding shares of our non-voting common stock, in each case, on a one-for-one basis, into an aggregate of 32,477,209 shares of our common stock (after giving effect to the Reverse Stock Split), which was effective on October 31, 2023, (iii) the creation and issuance of 2,500 shares of our Series C Preferred Stock (after giving effect to the Reverse Stock Split), which will occur prior to the effectiveness of the registration statement of which this prospectus forms a part, and (iv) the filing and effectiveness of our amended and restated certificate of incorporation and the adoption of our amended and restated bylaws, each of which will occur in connection with the effectiveness of the registration statement of which this prospectus forms a part. |
The share numbers in the table below have been adjusted to reflect the Reverse Stock Splits and the Capital Stock Adjustments.
This table should be read in conjunction with, and is qualified in its entirety by reference to, our financial statements and related notes appearing elsewhere in this prospectus.
| As of September 30, 2023 | ||||||||
| Actual | Pro Forma | |||||||
|
(unaudited) (in thousands, except for share and |
||||||||
| Cash and cash equivalents | $ | 10,766 | $ | 10,766 | ||||
| Stockholders’ equity/(deficit): | ||||||||
| Preferred Stock, $0.00001 par value per share; 20,000,000 total shares authorized, actual and pro forma; 8,750,000 Series A Preferred shares authorized, issued and outstanding, actual; no Series A Preferred shares authorized, issued and outstanding, pro forma | — | — | ||||||
| Preferred Stock, $0.00001 par value per share; 20,000,000 total shares authorized, actual and pro forma; 5,000,000 Series B Preferred shares authorized, actual; 4,171,445 Series B Preferred shares issued and outstanding, actual; no Series B Preferred shares authorized, issued and outstanding, pro forma | — | — | ||||||
| Preferred Stock, $0.00001 par value per share; 20,000,000 total shares authorized, actual and pro forma; 5,000,000 Series B-1 Preferred shares authorized, actual; 74,922 Series B-1 Preferred shares issued and outstanding, actual; no Series B-1 Preferred shares authorized, issued and outstanding, pro forma | — | — | ||||||
| Preferred Stock, $0.00001 par value per share; 20,000,000 total shares authorized, actual and pro forma; no Series C Preferred shares authorized, issued and outstanding, actual; 2,500 Series C Preferred shares authorized, issued and outstanding, pro forma | — | — | ||||||
| Non-voting Common stock, $0.00001 par value per share; 30,000,000 shares authorized and 28,230,842 shares issued and outstanding, actual; no shares authorized, issued and outstanding, pro forma | 1 | — | ||||||
| Voting Common Stock, $0.00001 par value per share; 100,000,000 shares authorized, actual and pro forma; no shares issued or outstanding, actual; 32,477,209 shares issued and outstanding, pro forma | — | 1 | ||||||
| Additional paid-in capital | 25,177 | 25,177 | ||||||
| Accumulated deficit | (14,640 | ) | (14,640 | ) | ||||
| Total stockholders’ equity | $ | 10,538 | $ | 10,538 | ||||
| Total capitalization | $ | 10,538 | $ | 10,538 | ||||
The number of shares of our voting common stock reflected in our actual and pro forma information set forth in the table above excludes:
| ● | 3,788,500 shares of common stock issuable upon exercise of stock options outstanding under our 2022 Stock Plan (as defined herein) as of September 30, 2023, with a weighted-average exercise price of $2.36 per share; | |
| ● | 8,711,500 shares of common stock reserved for issuance under our 2022 Stock Plan; | |
| ● | 14,859 shares of Series B-1 Preferred Stock issued after September 30, 2023, which upon Direct Listing will automatically convert to shares of common stock on a one-for-one basis; | |
| ● | 10,321 shares of common stock underlying warrants to be issued in connection with the issuance of certain shares of the Series B-1 Preferred Stock; and | |
| ● | Shares of common stock underlying warrants to purchase common stock that will be issued to certain investors upon the Direct Listing pursuant to the Share Subscription Agreement, which underlying shares of common stock shall be equal to 4% of our total equity interests outstanding immediately after consummation of the Direct Listing, as more particularly discussed under “Risk Factors—Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our product candidates on unfavorable terms to us.” |
| 65 |
MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and related notes and other financial information appearing elsewhere in this prospectus. Some of the information contained in this discussion and analysis or set forth elsewhere in this prospectus, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the “Risk Factors” section of this prospectus, our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a clinical-stage cell therapy company focused on developing and commercializing fibroblast-based therapies for patients suffering from chronic diseases with significant unmet medical needs, including degenerative disc disease, multiple sclerosis, wound healing, and certain cancers, and potential extension of life applications including thymic and splenic involution reversal. At present, our novel manufacturing process entails collecting excess tissue from surgical procedures and using the allogeneic fibroblasts to grow a cell bank for use in our procedures. Our most advanced product candidates are CybroCell™ and CYMS101.
CybroCell™ is an allogeneic fibroblast cell-based therapy for degenerative disc disease and is being designed as an alternative method for repairing the cartilage of the intervertebral disc (or any other articular cartilage). We have completed two animal studies. The results from the studies were positive and resulted in “first in human” trial approval. We have received IND clearance from the FDA, conditional upon approval of our master cell bank, to run a Phase 1/2 study for patients suffering from degenerative disc disease and will be conducting this study within the United States. A timeline will be determined through discussions with the FDA.
We are developing CYMS101 as an allogeneic fibroblast cell-based therapy to treat MS. After completing animal studies using CYMS101 (allogeneic fibroblast cells), we received approval to conduct clinical investigations in Mexico using the fibroblast cell composition for patients with MS and have completed a Phase 1 study. The study was conducted in five participants. The primary objective of the study was to assess safety, and the secondary objective was to assess efficacy. We are currently conducting further research to determine the mode of action of fibroblasts in oligodendrocyte expansion and expect to file an IND application for a Phase 2 clinical trial in MS. We will likely seek a strategic partner to collaborate with us on the development of CYMS101 either before initiating the Phase 2 study, or after its completion, if successful, and prior to commencing with a Phase 3 clinical trial.
| 66 |
We are in the early stages of developing CYWC628 as an allogeneic fibroblast cell-based therapy for wound healing. Our studies are presently focused on utilizing fibroblasts and fibroblast-derived cells to treat wounds in diabetic mice and rats. Based upon our results achieved to date, we plan to pursue an IND submission with the FDA for wound healing as early as 2024.
We also have cancer and extension of life programs in the early stages of development and we plan to accelerate such programs as funding allows.
We currently purchase our cell therapy product candidates from a CDMO. We are in the process of contracting with a CDMO for the transfer of our experimental cell bank to produce our master cell bank, working cell bank and our fibroblast cell-based product candidates to enable clinical trials. If our product candidates receive marketing approval, we will evaluate the longer-term feasibility of building our own cGMP manufacturing facility or continuing to outsource production to a CDMO for clinical testing and commercial sales.
Since our spinoff from FibroGenesis in April 2021, our operations have included business planning, hiring personnel, raising capital, building our intellectual property portfolio and performing research and development on our product candidates and our fibroblast technology, leveraging the clinical benefits of fibroblasts as the basis of our cell therapy platform.
We have incurred net losses since inception and expect to incur losses in the future as we continue our research and development activities. To date, we have funded our operations primarily through investment from FibroGenesis, the issuance of $5.6 million of our convertible promissory notes that were issued from December 2021 through April 2022, and the issuance of $18.6 million of preferred stock.
As of September 30, 2023, we had cash and cash equivalents of approximately $10.8 million. Since our inception, we have incurred significant operating losses. We incurred net losses of approximately $6.8 million and $3.6 million for the nine months ended September 30, 2023 and 2022, respectively, and $5.1 million and $1.6 million for the years ended December 31, 2022 and 2021, respectively. As of September 30, 2023, we had an accumulated deficit of approximately $14.6 million. We expect to continue to incur significant expenses and operating losses for the next several years. See “—Funding Requirements” below.
We expect to continue to incur significant losses for the foreseeable future, and we expect these losses to increase substantially if and as we:
| ● | advance the development of our lead product candidates through clinical development, and, if approved by the FDA, commercialization; |
| ● | advance our preclinical development programs into clinical development; |
| ● | incur manufacturing costs for cell production to supply our product candidates; |
| ● | seek regulatory approvals for any of our product candidates that successfully complete clinical trials; |
| ● | increase our research and development activities to identify and develop new product candidates; |
| ● | hire additional personnel; |
| ● | expand our operational, financial and management systems; |
| ● | meet the requirements and demands of being a public company; |
| ● | invest in further development to protect and expand our intellectual property; |
| ● | establish a sales, marketing, medical affairs and distribution infrastructure to commercialize any product candidates for which we may obtain marketing approval and intend to commercialize; and |
| ● | expand our manufacturing and develop our commercialization efforts. |
| 67 |
Due to the numerous risks and uncertainties associated with biopharmaceutical product development and the economic and developmental uncertainty, we may be unable to accurately predict the timing or magnitude of all expenses. Our ability to ultimately generate revenue to achieve profitability will depend heavily on the development, approval, and subsequent commercialization of our product candidates. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.
As a result, we will need substantial additional funding to support our long-term continuing operations and pursue our growth strategy. Until such time as we can generate significant revenue from product sales, if ever, we expect to finance our operations through the sale of equity, debt financings or other capital sources, which may include collaborations with other companies or other strategic transactions. We may not be able to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements as and when needed, we will have to significantly delay, reduce or eliminate the development and commercialization of one or more of our product candidates or delay our pursuit of potential in-licenses or acquisitions.
Components of Results of Operations
Revenue
To date, we have not generated any revenue from product sales and do not expect to generate any revenue from the sale of products in the foreseeable future. If our development efforts for any of our product candidates are successful and result in regulatory approval, we may generate revenue in the future from product sales. We cannot predict if, when or to what extent we will generate revenue from the commercialization and sale of any of our product candidates. We may never succeed in obtaining regulatory approval for any of our product candidates.
Research and Development Expenses
Our research and development expenses consist of expenses incurred in connection with the development of our product candidates and include:
| ● | employee-related expenses, which include salaries, benefits, travel and stock-based compensation for our research and development personnel; |
| ● | laboratory equipment and supplies; |
| ● | direct third-party costs such as expenses incurred under agreements with CROs and CMOs; |
| ● | consultants that conduct research and development activities on our behalf; |
| ● | costs associated with conducting preclinical studies and clinical trials; |
| ● | costs associated with technology; and |
| ● | facilities and other allocated expenses, which include expenses for rent and other facility related costs and other supplies. |
We expense research and development costs as incurred. Nonrefundable advance payments that we make for goods or services to be received in the future for use in research and development activities are recorded as prepaid expenses. The prepaid amounts are expensed as the related goods are delivered or the services are performed.
| 68 |
We expect our research and development expenses to increase substantially for the foreseeable future as we continue to invest in research and development activities related to developing our product candidates as they advance into later stages of clinical development and our other product candidates in preclinical development as they advance into clinical development. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming, and the successful development of our product candidates is highly uncertain. As a result, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenue from the commercialization and sale of any of our product candidates. This is due to the numerous risks and uncertainties associated with developing product candidates, including uncertainty related to:
| ● | the duration, costs and timing of clinical trials of our current development programs and any further clinical trials related to new product candidates; |
| ● | the sufficiency of our financial and other resources to complete the necessary preclinical studies and clinical trials; |
| ● | the acceptance of IND applications for future clinical trials; |
| ● | the successful and timely enrollment and completion of clinical trials; |
| ● | the successful completion of preclinical studies and clinical trials; |
| ● | successful data from our clinical program that supports an acceptable risk-benefit profile of our product candidates in the intended populations; |
| ● | the receipt and maintenance of regulatory and marketing approvals from applicable regulatory authorities; |
| ● | establishing agreements with third-party manufacturers for clinical supply for our clinical trials and commercial manufacturing, if any of our product candidates are approved; |
| ● | the entry into collaborations to further the development of our product candidates; |
| ● | obtaining and maintaining patent and trade secret protection and regulatory exclusivity for our product candidates; and |
| ● | successfully launching our product candidates and achieving commercial sales, if and when approved. |
A change in the outcome of any of these variables with respect to the development of any of our programs or any product candidate we develop would significantly change the costs, timing and viability associated with the development and/or regulatory approval of such programs or product candidates.
General, Administrative and Other Expenses
Our general, administrative, and other expenses consist primarily of personnel costs, allocated facilities costs, and other expenses for outside professional services, including legal, marketing, investor relations, human resources services, and accounting services. Personnel costs consist of salaries, benefits, and stock-based compensation for our general and administrative personnel. We expect to incur additional expenses as a result of operating as a public company, including expenses related to compliance with the rules and regulations of the SEC, Nasdaq, additional insurance expenses, investor relations activities and other administrative and professional services. We also expect to increase the size of our administrative function to support the growth of our business.
Interest Expense
Our interest expense consists primarily of accrued interest expense and amortization of discount on our convertible notes.
| 69 |
Statements of Operations
Results of Operations
Comparison of Fiscal Years December 31, 2022 and 2021
The following table sets forth our results of operations for the years ended December 31, 2022 and 2021.
| Fiscal Year Ended December 31, |
Change Amount | |||||||||||
| 2022 | 2021 | |||||||||||
| (in thousands) | ||||||||||||
| Operating expenses: | ||||||||||||
| Research and development | $ | 1,147 | $ | 521 | $ | 626 | ||||||
| General, administrative and other | 3,320 | 1,057 | 2,263 | |||||||||
| Total operating expenses | 4,467 | 1,578 | 2,889 | |||||||||
| Loss from operations | (4,467 | ) | (1,578 | ) | (2,889 | ) | ||||||
| Interest expense | (654 | ) | (4 | ) | (650 | ) | ||||||
| Net loss | $ | (5,121 | ) | $ | (1,582 | ) | $ | (3,539 | ) | |||
Research and Development Expenses
Research and development expenses were $1.1 million and $0.5 million for the years ended December 31, 2022 and 2021, respectively. The increase of $0.6 million was primarily due to:
| ● | increased personnel related expenses of $0.4 million due to hiring our chief scientific officer in July 2021 and two additional research scientists by December 2022; |
| ● | increased research facility costs of $0.1 million due to licensing and expanding temporary laboratory and office space; and |
| ● | increased research supplies of $0.1 million due to increased laboratory personnel and preclinical studies. |
Research and development expenses are not tracked by product candidate.
General, Administrative and Other Expenses
General, administrative and other expenses were $3.3 million and $1.1 million for the years ended December 31, 2022 and 2021, respectively. The increase of $2.3 million was primarily due to:
| ● | increased personnel-related expenses of $0.9 million due to the timing of hiring our chief executive officer in August 2021 and our chief financial officer in June 2022; |
| ● | increased accounting, legal, marketing and consulting expenses of $0.7 million for costs associated with preparing to become a public company; |
| ● | increased facilities expense of $0.4 million for the cost of our leased office space, and |
| ● | increased board of directors expenses of $0.2 million due to establishing an independent board of directors after formation in 2021. |
| 70 |
Interest Expense
Interest expense was $0.7 million and $0.0 million for the years ended December 31, 2022 and 2021, respectively. The increase of $0.7 million was due to the issuance of convertible notes in December 2021, January 2022, and April 2022. Interest expense was recorded in 2022 for the nominal interest rate of 6.0% plus the amortization of the discount on the 2022 convertible notes.
Income Taxes
The effective income tax rate was 0.0% for all periods. Currently, we have recorded a full valuation allowance against our net deferred tax assets.
Comparison of Nine Months Ended September 30, 2023 and 2022
The following table sets forth our results of operations for the nine months ended September 30, 2023 and 2022.
|
Nine Months Ended September 30, |
Change Amount | |||||||||||
| 2023 | 2022 | |||||||||||
| (unaudited, in thousands) | ||||||||||||
| Operating expenses: | ||||||||||||
| Research and development | $ | 1,595 | $ | 802 | $ | 793 | ||||||
| General, administrative and other | 4,814 | 2,361 | 2,453 | |||||||||
| Total operating expenses | 6,409 | 3,163 | 3,246 | |||||||||
| Loss from operations | (6,409 | ) | (3,163 | ) | (3,246 | ) | ||||||
| Other loss | (213 | ) | — | (213 | ) | |||||||
| Interest expense | (146 | ) | (434 | ) | 288 | |||||||
| Net loss | $ | (6,768 | ) | $ | (3,597 | ) | $ | (5,744 | ) | |||
Research and Development Expenses
Research and development expenses were $1.6 million and $0.8 million for the nine months ended September 30, 2023 and 2022, respectively. The increase of $0.8 million was primarily due to:
| ● | increased personnel related expenses of $0.5 million due to hiring three additional research scientists and granting stock options in 2023; |
| ● | increased research consultant costs of $0.1 million to provide technical writing support; and |
| ● | increased research facility and laboratory supplies of $0.2 million due to increased laboratory personnel and preclinical studies. |
Research and development expenses are not tracked by product candidate.
| 71 |
General, Administrative and Other Expenses
General, administrative and other expenses were $4.8 million and $2.4 million for the nine months ended September 30, 2023 and 2022, respectively. The increase of $2.4 million was primarily due to:
| ● | increased personnel-related expenses of $1.4 million due to the timing of hiring our chief financial officer in June 2022, stock options granted in 2023, and executive bonus in 2023; |
| ● | increased accounting, legal, marketing and consulting expenses of $0.5 million for costs associated with preparing to become a public company; |
| ● | increased facilities expense of $0.3 million for the cost of our leased office space, and |
| ● | increased board of directors expenses of $0.2 million due to stock options granted in 2023. |
Other loss
Other loss was $0.2 million and none for the nine months ended September 30, 2023 and 2022, respectively. Other loss is comprised of the ROFN payments to FibroGenesis in excess of the derivative liability established at inception of the ROFN Agreement in January 2023.
Interest Expense
Interest expense was $0.1 million and $0.4 million for the nine months ended September 30, 2023 and 2022, respectively. The decrease of $0.3 million was due to the maturities and conversions of the notes during the nine months ended September 30, 2023. Interest expense was recorded in 2022 and 2023 for the nominal interest rate of 6.0% plus the amortization of the discount on the 2022 convertible notes.
Income Taxes
The effective income tax rate was 0.0% for all periods. Currently, we have recorded a full valuation allowance against our net deferred tax assets.
Liquidity and Capital Resources
Overview
To date, we have financed our operations primarily with investment from FibroGenesis, proceeds from borrowings under our convertible loan agreements, and proceeds from the issuance of preferred stock. From inception through September 30, 2023, we have received aggregate proceeds of approximately $5.6 million from sales of our convertible notes and $18.3 million from the issuance of preferred stock. As of September 30, 2023, we had cash and cash equivalents of approximately $10.8 million and an accumulated deficit of approximately $14.6 million. As of September 30, 2023, we had no outstanding debt.
Cash Flows
The following table sets forth a summary of our cash flows for the years ended December 31, 2022 and 2021.
| Year Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| (in thousands) | ||||||||
| Net cash used in operating activities | $ | (4,066 | ) | $ | (1,410 | ) | ||
| Net cash provided by financing activities | 5,925 | 1,817 | ||||||
| Net increase in cash and cash equivalents | $ | 1,859 | $ | 407 | ||||
Operating Activities
Net cash used in operating activities was $4.1 million and $1.4 million for the years ended December 31, 2022 and 2021, respectively, and consisted primarily of net losses of $5.1 million and $1.6 million, respectively. An increase of $0.5 million in accounts payable and accrued expenses, plus noncash expenses of $0.3 million in stock-based compensation expense and $0.4 million in amortization of convertible notes debt discount, partially offset the net losses in the year ended December 31, 2022. An increase of $0.2 million in accounts payable and accrued expenses partially offset the net losses in the year ended December 31, 2021.
| 72 |
Financing Activities
Net cash provided by financing activities was approximately $5.9 million and $1.8 million for the years ended December 31, 2022 and 2021. In 2021, we received nearly $1.0 million from FibroGenesis through a loan agreement and repaid approximately $0.8 million in the same year, and FibroGenesis invested $0.3 million during the first quarter of 2021 as part of our carve-out from FibroGenesis. We also received $1.3 million from the issuance of convertible notes in December 2021. In 2022, we received $4.3 million from the issuance of convertible notes in January and April 2022, and received $2.2 million from the issuance of preferred stock in December 2022. We repaid $0.2 million and loaned $0.4 million to FibroGenesis in 2022, and FibroGenesis repaid $0.1 million to us in 2022.
The following table sets forth a summary of our cash flows for the nine months ended September 30, 2023 and 2022.
| Nine Months Ended September 30, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Net cash used in operating activities | $ | (4,800 | ) | $ | (2,893 | ) | ||
| Net cash used in investing activities | (493 | ) | — | |||||
| Net cash provided by financing activities | 13,793 | 3,775 | ||||||
| Net increase in cash and cash equivalents | $ | 8,500 | $ | 882 | ||||
Operating Activities
Net cash used in operating activities was $4.8 million and $2.9 million for the nine months ended September 30, 2023 and 2022, respectively, and consisted primarily of net losses of $6.8 million and $3.6 million, respectively. An increase of $0.4 million in accounts payable and accrued expenses, plus noncash expenses of $1.3 million in stock-based compensation expense,a $0.2 million in loss on derivative liability, and $0.1 million in amortization of convertible notes debt discount partially offset the net losses in the nine months ended September 30, 2023. An increase of $0.3 million in accounts payable and accrued expenses, plus noncash expenses of $0.2 million in stock-based compensation expense and $0.2 million in amortization of convertible notes debt discount partially offset the net losses in the nine months ended September 30, 2022.
Investing Activities
Net cash used in investing activities was approximately $0.5 million and none for the nine months ended September 30, 2023 and 2022. The increase was due to the purchase of laboratory equipment.
Financing Activities
Net cash provided by financing activities was approximately $13.8 million and $3.8 million for the nine months ended September 30, 2023 and 2022. We received from FibroGenesis repayment of a $0.3 million loan, paid to FibroGenesis $2.6 million under the ROFN Agreement (as defined herein), and we received $16.1 million in total from the issuance of Series B Preferred Stock and Series B-1 Preferred Stock during the nine months ended September 30, 2023. We repaid to FibroGenesis a $0.2 million loan, loaned $0.3 million to FibroGenesis and received $4.3 million from the issuance of convertible notes during the nine months ended September 30, 2022.
Funding Requirements
We do not have any products approved for sale, and we have never generated any revenue from contracts with customers. We do not expect to generate any meaningful revenue unless and until we obtain regulatory approval of and commercialize any of our current or future product candidates and we do not know when, or if, that will occur. We expect to continue to incur significant losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our current and future product candidates, and begin to commercialize any approved products. We are subject to all the risks typically related to the development of new product candidates, and we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. Moreover, following the completion of this offering, we expect to incur additional costs associated with operating as a public company.
The financial statements have been prepared as though we will continue as a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. We have incurred operating losses and negative cash flows from operations since inception. As of September 30, 2023, we had an accumulated deficit of approximately $14.6 million. Management expects to continue to incur operating losses and negative cash flows.
We will need to raise additional capital to continue to fund our operations. We believe we will be able to obtain additional capital through equity financings or other arrangements to fund operations; however, there can be no assurance that such additional financing, if available, can be obtained on acceptable terms. If we are unable to obtain such additional financing, future operations would need to be scaled back or discontinued.
We believe that our existing capital will enable us to fund our operations through at least December 31, 2024. We may need to raise additional capital in connection with our cash needs for capital expenditures and working capital beyond December 31, 2024. We have based the foregoing estimate on assumptions that may prove to be incorrect, and we could use our capital resources sooner than we expect.
Our future funding requirements will depend on many factors, including, but not limited to:
| ● | the initiation, progress, timeline, cost and results of our clinical trials for our product candidates; |
| ● | the initiation, progress, timeline, cost and results of additional research and preclinical studies related to pipeline development and other research programs we initiate in the future; |
| ● | the cost and timing of manufacturing activities, including our planned manufacturing scale-up activities associated with our product candidates and other programs as we advance them through preclinical and clinical development through commercialization; |
| ● | the potential expansion of our current development programs to seek new indications; |
| ● | the outcome, timing and cost of meeting regulatory requirements established by the FDA and other comparable foreign regulatory authorities; |
| 73 |
| ● | the cost of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights, in-licensed or otherwise; |
| ● | the effect of competing technological and market developments; |
| ● | the payment of licensing fees, potential royalty payments and potential milestone payments; |
| ● | the cost of general operating expenses; |
| ● | the cost of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval in regions where we choose to commercialize our products on our own; and |
| ● | the costs of operating as a public company. |
Further, our operating plan may change, and we may need additional funds to meet operational needs and capital requirements for clinical trials and other research and development expenditures.
If we need to raise additional capital to fund our operations, funding may not be available to us on acceptable terms, or at all. If we are unable to obtain adequate financing when needed, we may have to delay, reduce the scope of or suspend one or more of our preclinical studies, clinical trials, research and development programs or commercialization efforts. We may seek to raise any necessary additional capital through a combination of public or private equity offerings, debt financings, collaborations and other licensing arrangements. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional capital through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish certain valuable rights to our product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable to us.
Contractual Obligations and Commitments
As of December 31, 2022, our commitments consisted of convertible notes payable and two leases for laboratory and office space in Houston, Texas. As of September 30, 2023, our commitments consisted of two leases for laboratory and office space in Houston, Texas.
The following table summarizes our contractual obligations as of December 31, 2022.
| Total | Less Than One Year |
1–3 Years |
3–5 Years |
More Than 5 Years |
||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Convertible notes payable obligations | $ | 5,600 | $ | 5,600 | $ | — | $ | — | $ | — | ||||||||||
| Operating lease obligations | 2,484 | 466 | 1,509 | 509 | — | |||||||||||||||
| Total | $ | 8,084 | $ | 6,066 | $ | 1,509 | $ | 509 | $ | — | ||||||||||
The convertible notes payable were converted into Series B Preferred Stock during the nine months ended September 30, 2023.
The following table summarizes our contractual obligations as of September 30, 2023.
| Payments Due by Period | ||||||||||||||||||||
| Total | Less
Than One Year |
1–3 Years |
3–5 Years |
More
Than 5 Years |
||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Operating lease obligations | $ | 2,104 | $ | 475 | $ | 982 | $ | 647 | $ | — | ||||||||||
| Total | $ | 2,104 | $ | 475 | $ | 982 | $ | 647 | $ | — | ||||||||||
Off-Balance Sheet Arrangements
During the periods presented, we did not have, nor do we currently have, any off-balance sheet arrangements or holdings in any variable interest entities.
| 74 |
Critical Accounting Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with GAAP. The preparation of these financial statements requires us to make estimates, judgments and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities as of the dates of the balance sheets and the reported amounts of expenses during the reporting periods. In accordance with GAAP, we evaluate our estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We define our critical accounting estimates as those under GAAP that require us to make subjective estimates and judgments about matters that are uncertain and are likely to have a material impact on our financial condition and results of operations, as well as the specific manner in which we apply those principles. While our significant accounting policies are more fully described in Note 2 to our financial statements included elsewhere in this prospectus, we believe the following are the critical accounting estimates used in the preparation of our financial statements that require significant estimates and judgments.
Net
Parent Investment
Because our financial statements prior to inception on April 8, 2021, are derived from the historical records of FibroGenesis, the net parent investment is presented within stockholders’ deficit on the balance sheets. As a then subsidiary of FibroGenesis, we were dependent upon FibroGenesis for all of our working capital and financing requirements prior to entering into our convertible note agreements. Financial transactions that relate to FibroBiologics but occurred at the parent level are accounted for through the net parent investment account. Accordingly, none of FibroGenesis’ cash, cash equivalents or debt have been assigned to us in the financial statements. Net parent investment represents FibroGenesis’ interest in our recorded net assets.
Research and Development
Research and development costs are charged to expense as incurred. Research and development costs consist of costs incurred in performing research and development activities, including salaries and bonuses, scientist recruiting costs, employee benefits, facilities costs, laboratory supplies, manufacturing expenses, preclinical expenses, research materials, and consulting and other contracted services. Costs for certain research and development activities are recognized based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the financial statements as prepaid or accrued research and development.
Convertible Notes Payable
We entered into multiple convertible promissory note agreements in December 2021, or, collectively, the 2021 Notes. Under the 2021 Notes, we received $1.3 million, which accrued simple interest at a rate of 6.0% per annum and was to mature in the event of our initial public offering. Upon maturity of the 2021 Notes, the holders could elect to receive cash payment in full for the outstanding principal and interest or elect a one-year extension at the discretion of the holders of the 2021 Notes.
In the event that we issued and sold shares of our capital stock in excess of $10.0 million, the outstanding balance of the 2021 Notes and accrued interest could be converted into a fixed number of shares of common stock, subject only to customary anti-dilution provisions for any recapitalization that may occur.
Based on the terms of the 2021 Notes, we evaluated the conversion option feature in accordance with ASC 815 Derivatives and Hedging. It provides three criteria that, if met, require companies to bifurcate conversion options from their host instruments and account for them as freestanding derivative financial instruments. These three criteria include circumstances in which (i) the economic characteristics and risks of the embedded derivative instrument are not clearly and closely related to the economic characteristics and risks of the host contract, (ii) the hybrid instrument that embodies both the embedded derivative instrument and the host contract is not re-measured at fair value under otherwise applicable generally accepted accounting principles with changes in fair value reported in earnings as they occur and (iii) a separate instrument with the same terms as the embedded derivative instrument would be considered a derivative instrument.
| 75 |
At the issuance of the 2021 Notes, and at December 31, 2021 and 2022, we determined that an embedded derivative for the conversion feature did not meet the criteria because it met the “indexed to the entity’s own stock” exception and therefore was not required to be bifurcated from the host instrument. As noted below, all of the 2021 Notes were converted into shares of our Series B Preferred Stock during the nine months ended September 30, 2023.
We issued additional convertible promissory notes between January and April 2022 with a total principal amount of $4.3 million and a one-year maturity, or, collectively, the 2022 Notes. The 2022 Notes could be converted at the lesser of (i) a 15% discount to the offering price of our common stock in the event of our initial public offering or (ii) the quotient of $200.0 million divided by total equity interests prior to the dilution from the offering. The conversion option feature in the 2022 Notes was evaluated in accordance with ASC 815, and a derivative liability for the $0.5 million estimated fair value of the conversion option was recorded at the time the notes were issued and as of December 31, 2022. An offsetting discount on the issuance of the notes was recorded and is being amortized to interest expense over the expected life of the 2022 Notes.
In February 2023, we converted the principal and interest on $3.7 million of principal value of the 2022 Notes into the equivalent of 799,603 shares of our Series B Preferred Stock. In April 2023, we converted the principal and interest on $1.6 million of principal value of the 2021 Notes and $0.3 million of principal value on the 2022 Notes into the equivalent of 353,713 shares of our Series B Preferred Stock. In June 2023, we converted the remaining $0.3 million of principal value on the 2022 Notes into the equivalent of 66,077 shares of our Series B Preferred Stock. There was no outstanding principal balance on the 2022 Notes at September 30, 2022.
The convertible debt balances were none and $5.6 million at September 30, 2023 and December 31, 2022, respectively.
Stock-Based Compensation
We measure all stock option grants to employees, directors and non-employees based on their fair value on the date of the grant and recognize the corresponding compensation expense of those awards using the straight-line method over the requisite service period, which is generally the vesting period of the respective award. Forfeitures are accounted for as they occur.
We classify stock-based compensation expense in our statements of operations in the same way the award recipient’s payroll costs are classified or in which the award recipient’s service payments are classified.
We estimate the fair value of each stock option grant using the Black-Scholes option-pricing model, which uses as inputs the fair value of our common stock and assumptions we make for the volatility of our common stock, the expected term of our stock options, the risk-free interest rate for a period that approximates the expected term of our stock options and our expected dividend yield.
The estimated fair value of our common stock underlying our stock-based awards has been determined by our board of directors as of each option grant date with input from management, considering our most recently available third-party valuations of common stock and our board of directors’ assessment of additional objective and subjective factors that it believed were relevant and which may have changed from the date of the most recent valuation through the date of the grant. These third-party valuations were performed in accordance with the guidance outlined in the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation (the Practice Aid).
Once a public trading market for our common stock has been established in connection with the completion of the Direct Listing, it will no longer be necessary for our board of directors to estimate the fair value of our common stock in connection with our accounting for granted stock options and other such awards we may grant, as the fair value of our common stock will be determined based on the quoted market price of our common stock.
See Note 11 to our audited financial statements included elsewhere in this prospectus for further information concerning the assumptions we used in in determining stock-based compensation.
ROFN Agreement
In January 2023, we entered into an Agreement Regarding Right of First Negotiation with FibroGenesis, or the ROFN Agreement. In exchange for FibroGenesis’ consent to amend our certificate of incorporation to (i) eliminate upon our underwritten initial public offering or the direct listing of our common stock on a securities exchange (which we collectively refer to as an IPO) or sale of our company, the liquidation preference for the Series A Preferred Stock, (ii) make the Series B Preferred Stock liquidation preference equal to Series A Preferred Stock, and (iii) to provide that upon an IPO or sale of our company, the Series A Preferred Stock will be canceled for no consideration, we agreed to pay to FibroGenesis 15% of the gross proceeds from any equity investments in us prior to an IPO or sale of our company. In addition, we received a five-year right of first negotiation if FibroGenesis decides to license externally any of its technology. Based upon our management’s estimates at execution of the ROFN Agreement of capital to be raised in advance of a public listing, we recorded a derivative liability of $2.6 million for the expected future payments to FibroGenesis. As a deemed dividend, the derivative liability was recorded first against the net Parent Investment and then to Additional paid-in capital after the net Parent Investment was eliminated. Amounts paid to FibroGenesis in excess of the derivative liability are recorded as other losses in the statement of operations. The deemed dividend is included as a reduction to net loss in the calculation of amount available to common stockholders in determining earnings per share.
The JOBS Act
We are an “emerging growth company” as defined in the JOBS Act. Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies.
We have elected to use this extended transition period for complying with new or revised accounting standards that have different effective dates for public and private companies until the earlier of the date we (i) are no longer an emerging growth company or (ii) affirmatively and irrevocably opt out of the extended transition period provided in the JOBS Act. As a result, our financial statements may not be comparable to companies that comply with new or revised accounting pronouncements as of public company effective dates. If we were to subsequently elect instead to comply with these public company effective dates, such election would be irrevocable pursuant to the JOBS Act.
We will remain an emerging growth company until the earliest of (i) December 31, 2028, (ii) the last day of the fiscal year in which we have total annual gross revenue of at least $1.235 billion, (iii) the last day of the fiscal year in which we are deemed to be a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our common stock held by non-affiliates was $700.0 million or more as of the last business day of the second fiscal quarter of such year or (iv) the date on which we have issued more than $1.0 billion in non-convertible debt securities during the prior three-year period.
Quantitative and Qualitative Disclosures About Market Risk
The primary objectives of our investment activities are to ensure liquidity and to preserve capital. We are exposed to market risks in the ordinary course of our business. These risks primarily include interest rate sensitivities. We had cash and cash equivalents of approximately $10.8 million and $2.3 million as of September 30, 2023 and December 31, 2022, respectively, which consisted of bank deposits. Historical fluctuations in interest rates have not been significant for us. Due to the short-term maturities of our cash equivalents, an immediate 100 basis point change in interest rates would not have a material effect on the fair market value of our cash equivalents. We do not believe that inflation, interest rate changes, or foreign currency exchange rate fluctuations had a significant impact on our results of operations for any periods presented herein.
| 76 |
Overview
We are a clinical-stage cell therapy company focused on developing and commercializing fibroblast-based therapies for patients suffering from chronic diseases with significant unmet medical needs, including degenerative disc disease, multiple sclerosis, wound healing, and certain cancers, and potential extension of life applications including thymic and splenic involution reversal.
We were formed in April 2021 as a Texas limited liability company under the name FibroBiologics, LLC, and converted to a Delaware corporation in December 2021 under the name Fibrobiologics, Inc. On April 14, 2023, we changed our name to FibroBiologics, Inc. In connection with our formation, we issued shares of our Series A Preferred Stock, or the Series A Preferred Stock, to our then parent, FibroGenesis, in return for rights to certain intellectual property through a patent assignment agreement and an intellectual property cross-licensing agreement. Developing the intellectual property obtained from FibroGenesis was the basis for our formation. Prior to our inception, preclinical research and development related to the aforementioned disease pathways took place under FibroGenesis.
Fibroblasts Technology Platform
Fibroblasts and stem cells are the only two cell types in the human body that can regenerate tissue and organs. Studies have indicated that mesenchymal stem cells and fibroblasts share many surface markers in common, and can differentiate into many cells including adipocytes, chondrocytes, osteoblasts, hepatocytes, and cardiomyocytes, and can regulate the immune system. However, transcriptomic and epigenetic studies have indicated a clear difference between the two cell types.
Fibroblasts comprise the main cell type of connective tissue, possessing a spindle-shaped morphology, whose classical function has historically been believed to produce an extracellular matrix responsible for maintaining the structural integrity of the tissue. Fibroblasts also play an important role in the proliferative phase of wound healing, resulting in the deposition of the extracellular matrix.
Fibroblasts are favorable to stem cells as a cell therapy treatment platform because fibroblasts:
| ● | can be non-invasively harvested from a variety of skin donors from surgical procedures such as tummy tuck flaps; |
| ● | have a faster doubling time in culture than stem cells; |
| ● | possess superior immune modulatory activity compared with stem cells; |
| ● | exhibit enhanced ability to produce regenerative cytokines and growth factors compared with stem cells; and |
| ● | are more economical to isolate, culture and expand compared with stem cells because fibroblasts do not require the use of expensive tissue culture media. |
| 77 |
Studies have demonstrated that allogeneic fibroblasts, much like mesenchymal stem cells, are immune-privileged and do not provoke an immune response in vitro and in vivo. These studies include that of Valente and colleagues (PMID 7646145) in which they looked at the aortic valve after heart transplantation and noted that even acute cases of acute myocardial rejection did not appear to compromise the long-term viability and durability of the valve, and the tissue viability was histologically confirmed and showed perfectly preserved fibroblasts1. In another study by O’Brien and colleagues (PMID 3682851) the researchers illustrated, using chromosomal analysis, long-term viability of the male donor fibroblast cells from a valve leaflet removed nine years after implantation into a female recipient. This illustrated that donor fibroblast cells are able to survive and proliferate in the host with destruction by the immune system2. If autologous fibroblasts were required instead, it would mean that cells would have to be harvested from each patient, processed and cultured, and then administered to the same patient, which would be more costly and inefficient. Because allogeneic fibroblasts do not cause an immune response, we are planning to build our own cGMP manufacturing facility to source allogeneic fibroblast cells for clinical testing of our product candidates and for commercial sales if product candidates receive marketing approval.
Our Strategy
We are leveraging fibroblast cells as a technology platform to research and develop innovative treatments for chronic diseases with significant unmet treatment needs. Our vision is to become a world leader in regenerative medicine through a rigorous scientific process and commitment to serving patients’ needs. To achieve our vision, we will focus our efforts on the following strategy:
| ● | Attract and retain scientists with the skill sets required to conduct preclinical studies and identify the optimal paths forward to clinical trials. | |
| ● | Prioritize our initial clinical development efforts on product candidates with the combination of significant unmet treatment needs, lower risk and market potential. | |
| ● | Partner with CROs with the relevant expertise and experience to successfully and timely execute clinical trials to generate reliable pivotal data that can be used to seek approvals. | |
| ● | Invest in critical capabilities required to produce and supply fibroblasts for clinical trials and initial commercialization. | |
| ● | Protect, expand and defend our intellectual property portfolio around fibroblasts. | |
| ● | Expand development efforts in product candidates with longer development timelines, greater risk and significant unmet treatment needs as funding allows. |
As of September 30, 2023, we had cash and cash equivalents of approximately $10.8 million. To advance our aforementioned strategy over the next 12 months, in addition to ongoing expenditures for personnel and infrastructure, the following research and development initiatives are expected to be funded with available capital:
| ● | Approximately $0.5 million to $0.7 million in capital expenditures for laboratory equipment to enable continued research and expand capabilities; | |
| ● | Approximately $0.3 million to $0.4 million to acquire clinical skin samples; | |
| ● | Approximately $0.2 million to $0.3 million for tissue and fibroblast cell characterization and optimization; | |
| ● | Approximately $0.2 million to $0.4 million to develop a master cell bank; | |
| ● | Approximately $0.1 million to $0.3 million to develop a working cell bank; | |
| ● | Approximately $0.1 million to $0.3 million to develop processes for transfer to a CDMO; | |
| ● | Approximately $0.1 million to $0.3 million to complete one additional animal study for CYWC628 in wound healing; | |
| ● | Approximately $0.1 million to submit an IND application for CYWC628 for a phase 1/2 clinical trial in wound healing; | |
| ● | Approximately $0.2 million to $0.3 million to complete a preclinical animal study to determine mode of action for CYMS101 in MS prior to submitting an IND application for a phase 1/2 clinical trial in MS; and | |
| ● | Approximately $0.1 million to initiate a preclinical animal study in CYTER915 for thymic involution reversal. |
We believe our available capital will allow us to develop the master cell bank, and working cell bank for transfer to a CDMO that will ultimately manufacture our drug products required to initiate phase 1/2 clinical trials for CybroCell, CYMS101, and CYWC628.
The above estimates are preliminary and subject to change. We cannot specify with certainty all of the particular uses for our available capital within the next 12 months. Due to uncertainties inherent in the development process, it is difficult to estimate the exact amounts of our available capital that will be used for any particular purpose. In addition, the amount, allocation and timing of our actual expenditures will depend upon numerous factors, including the results of our research and development efforts.
Our People
We have assembled an executive leadership team comprised of our founder, chief executive officer and chairperson of our board of directors, our chief scientific officer, and our chief financial officer, with successful track records in startup entrepreneurial companies and in the life sciences industry. Our executive leadership team works under the oversight of our board of directors who are recognized leaders with hands-on industry experience. We also have a team of world-renowned scientists with relevant expertise on our scientific advisory board to help guide our research and development efforts.
1 Source: Valente M, Faggian G, Billingham ME, Talenti E, Calabrese F, Casula R, Shumway NE, Thiene G. The aortic valve after heart transplantation. Ann Thorac Surg. 1995 Aug;60(2 Suppl):S135-40. doi: 10.1016/0003-4975(95)00251-f. PMID: 7646145.
2 Source: O’Brien MF, Stafford EG, Gardner MA, Pohlner PG, McGiffin DC. A comparison of aortic valve replacement with viable cryopreserved and fresh allograft valves, with a note on chromosomal studies. J Thorac Cardiovasc Surg. 1987 Dec;94(6):812-23. PMID: 3682851.
| 78 |
Our Current Pipeline
We have a pipeline of product candidates at various stages of development, including the following:
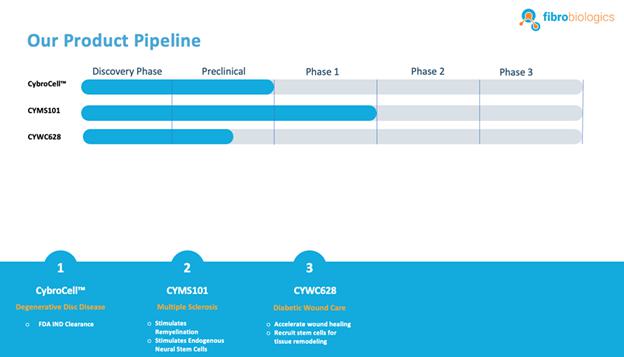
CybroCellTM for Degenerative Disc Disease
Degenerative Disc Disease
Back pain is strongly associated with degeneration of the intervertebral disc. Disc degeneration, although in many cases asymptomatic, is also associated with sciatica and disc herniation, pain or prolapse. It alters disc height and the mechanics of the rest of the spinal column, adversely affecting the behavior of other spinal structures such as muscles and ligaments. In the long term, it can lead to spinal stenosis, a major cause of pain and disability in the elderly. Its incidence is rising with current demographic changes and an increased aged population.
The disc acts as a joint between two vertebra and performs the following critical functions:
| ● | absorbs shock; | |
| ● | maintains motion; and | |
| ● | keeps stability. |
Discs degenerate far earlier than do other musculoskeletal tissues. The first unequivocal findings of degeneration in the lumbar discs are seen in the age group 11–16 years. About 20% of people in their teens have discs with mild signs of degeneration. The percentage increases sharply with age, particularly in males, so that around 10% of 50-year-old discs and 60% of 70-year-old discs are severely degenerated (Current Epidemiology of Low Back Pain” by Mattiuzzi et al, in 2020).
| 79 |
During growth and skeletal maturation, the boundary between annulus and nucleus becomes less obvious and, with increasing age, the nucleus generally becomes more fibrotic and less gel-like. With increasing age and degeneration, the disc changes in morphology, becoming more and more disorganized. Often, the annular lamellae becomes irregular, bifurcating and interdigitating, and the collagen and elastin networks also appear to become more disorganized.
Cleft formation with fissures frequently forms within the disc, particularly in the nucleus. Nerves and blood vessels are increasingly found with degeneration. Cell proliferation occurs, leading to cluster formation in the nucleus. Cell death also occurs, with the presence of cells with necrotic and apoptotic appearance. It has been reported that more than 50% of cells in adult discs are necrotic. With increasing age comes an increased incidence of degenerative changes, including cell death, cell proliferation, mucous degeneration, granular change and concentric tears. It is difficult to differentiate changes that occur solely due to aging from those that might be considered ‘pathological’.
According to research published in the Global Spine Journal, titled “Degenerative Lumbar Spine Disease: Estimating Global Incidence and Worldwide” by Ravindra et al published in 2018, approximately 266.0 million individuals suffer from degenerative spinal disease and lower back pain each year. In addition, 403.0 million individuals present annually with symptomatic disc degeneration, 103.0 million with spinal stenosis and 39.0 million with spondylolisthesis. Furthermore, lower back pain is considered as one of the chief complaints that may indicate an underlying spine-related disorder. According to the research published in the Journal of Hospital Management and Health Policy, titled “Current Epidemiology of Low Back Pain” by Mattiuzzi et al, in 2020, incidence, prevalence and disability-adjusted life years, or DALYs, of lower back pain are 245.9 million cases per year (15th worldwide cause), 577.0 million cases (15th worldwide cause) and 64.9 million (6th worldwide cause), respectively. The paper further stated that the risk of lower back pain is marginally higher in women compared to men. Chronic lower back pain is one of the common complaints that may indicate an underlying serious spinal disorder.
These statistics indicate the significant impact degenerative spine disorders can have on patients’ lives. These indications are associated with a diverse range of clinical symptoms such as weakness, low extremity pain and back pain, and can result in a significant reduction in the quality of life. The treatments used presently are mainly conservative and palliative and are aimed at returning patients to work. They range from bed rest (no longer recommended) to analgesia, the use of muscle relaxants or injection of corticosteroids, or local anesthetic and manipulation therapies. Various interventions (e.g. intradiscal electrotherapy) are also used, but despite anecdotal statements of success, trials thus far have found their use to be of little direct benefit. Disc degeneration-related pain may also be treated surgically either by artificial disc replacement or by immobilization of the affected vertebrae.
| 80 |
Available Treatments for Degenerative Disc Disease
Most patients suffering from degenerative disc disease, at least initially, show improvement with non-surgical interventions such as physical therapy, core strengthening, and stretching. When those interventions no longer provide relief, patients typically use therapeutics, which include conventional drugs such as opioids, non-steroidal anti-inflammatory drugs, and corticosteroids for pain relief. When these non-surgical therapeutics are no longer effective, patients may undergo surgical treatment, including the use of medical devices or implants, to provide relief.
The original surgical treatment for correcting degenerated disc is either to perform a discectomy or spinal fusion. Discectomy is an appropriate procedure and is routinely performed to remove the degenerated nucleus through a fenestration within the annulus. It allows removal of both the extruded nucleus (herniectomy) and the degenerated remaining inter-vertebral nucleus fragments. Although this procedure is ideal for decompressing and relieving the nervous system (root or cauda equina), it is a poor operation for the spine, due to its resulting disabling condition which leads to a degenerative cascade and may require an additional invasive surgical procedure, like fusion or arthroplasty. Discectomy brings a good short-term effect in relieving radicular pain, but it causes disc height reduction with neuro-foramen stenosis, instability of the treated level, poor result on back pain, and/or complications, such as spinal stenosis or facet pain.
Patients who undergo these procedures are usually on painkillers for weeks and have at least three to six months of recovery time. Therefore, there is a need for a less painful, less invasive and more effective method. The pitfalls of original treatment procedures have led to a search for the development of non-fusion technologies, such as disc or disc nucleus prosthesis. Disc arthroplasty with an artificial disc is an emerging treatment for patients with disc degeneration. Its advantages are to maintain motion, decrease incidence of adjacent segment degeneration, avoid complications related to fusion and allow early return to function. Currently, two kinds of devices are marketed: the total disc replacement and the nuclear replacement. However, both of these devices have major pitfalls.
There has been a growing demand for spinal artificial discs in the market globally. These devices are gaining popularity as they are designed with the intent to provide stabilization and eliminate pain while preserving motion of the functional spinal unit. In July 2021, Aesculap Implant Systems, LLC announced the long-term reporting from its pivotal trial for the activL® Artificial Disc.

Centinel Spine’s prodiscL is a Total Disc Replacement, or TDR, technology platform that offers a surgical alternative to fusion to qualified patients suffering from disc degeneration in the cervical and lumbar spine. ProdiscL implants are intended to relieve pain while allowing the potential for motion at the diseased spinal segment.

The activL Artificial Disc for one-level lumbar use is a weight-bearing modular implant consisting of two endplates and one polyethylene inlay and is intended as an alternative to fusion. It’s designed to allow controlled motion at the surgery level.
| 81 |
Total disc replacement is a bulky metallic prosthesis designed to replace the entire disc: annulus, nucleus and endplates. These prostheses use an invasive anterior (trans- or retro-peritoneal) approach that requires the presence of a vascular surgeon. Dislodgements, wear debris, degeneration of adjacent intervertebral discs, facet joint arthrosis and subsidence of this type of prosthesis have been reported. The artificial nucleus substitute preserves the remaining disc tissues and their functions. Its design allows its implantation through a posterior approach, but the major limitation of such nucleus prosthesis is that it can be used only in patients in whom disc degeneration is at an early or intermediate stage, because it requires the presence of a competent natural annulus. As a hydrogel-based device, it is fragile, and so does not resist the outstanding biomechanical constraints of the lumbar spine (shear forces). As inert materials, they may lose their mechanical properties over time, and tears and breakages have been reported. Replacing the nucleus only and leaving in place a damaged annulus generates the conditions for implant extrusion or recidivism of discal herniation.
In addition to disc replacements, there are current treatment options for tissue engineering and regenerative medicine, which represent new options for the treatment of degenerative disc disease. A variety of approaches are used to regenerate tissues. These approaches can be categorized into the following three groups:
| (i) | Biomaterials, without additional cells, that are used to send signals to attract cells and promote regeneration; | |
| (ii) | Cells alone may be used, to form a tissue; and | |
| (iii) | Cells may be used with a biomaterial scaffold that acts as a frame for developing tissues. |
While Autologous Chondrocyte Transplantation, or ACT, has been used for a few years to repair articular cartilage, tissue engineering for disc repair remains in its infancy. Intensive research is currently underway, and animal studies have shown the feasibility of tissue-engineered intervertebral disc. Typically, articular cartilage is a tissue that is not naturally regenerated once damaged. Recently, efforts have been made to reconstruct damaged biological tissues by regenerating a portion of the damaged tissues in laboratories. This approach, defined as “tissue engineering,” has received tremendous attention.
Tissue engineering involves the development of biocompatible materials capable of specifically interacting with biological tissues to produce functional tissue equivalents. Tissue engineering has a basic concept of collecting a desired tissue from a patient, isolating cells from the tissue specimen, proliferating cells, seeding the proliferated cells onto a biodegradable polymeric scaffold, culturing the cells for a predetermined period in vitro, and transplanting back the cell/polymer construct into the patient. More interestingly, recent pilot clinical trials have shown that ACT is an efficient treatment of herniated disc. The main disadvantage of ACT for disc repair is that it requires a disc biopsy. Therefore, there is a need for an improved method to restore disc anatomy and improve its functioning, and there remains a need for an improved method of cartilage repair.
Our Solution
CybroCell™ is an allogeneic fibroblast cell-based therapy for degenerative disc disease This new technology is being designed as an alternative method for repairing the cartilage of the intervertebral disc (or any other articular cartilage). The method is based on using Human Dermal Fibroblasts, or HDFs, which are forced to differentiate into chondrocyte-like cells in vivo using the mechanical force and intermittent hydrostatic pressure found in the spine, for chondrogenic differentiation of fibroblasts. We believe our solution will prove superior to existing treatments because it is less invasive, regenerates the disc, restores function and reduces pain, without debilitating long-term effects. We received IND clearance from the FDA on November 7, 2018, with an IND number of 18151. The trial is designed to assess the safety and efficacy of CybroCell™ administered through injection directly into a damaged intervertebral disc. The trial will enroll up to 15 participants with a primary outcome of safety and a secondary outcome of efficacy. Safety will be assessed as the occurrence/frequency of adverse events during the study procedures and for up to 12 months afterwards and will be recorded regardless of severity or relatedness to treatment. Serious adverse events will be documented throughout the 12-month follow-up period. The incidence and nature of adverse events will be tabulated and analyzed at baseline, pre, and post procedure including the three, six, and 12-months evaluations. These include complete physical exam (including vital signs of blood pressure, temperature, and heart rate), laboratory determinations (including urinalysis, hematology, and biochemistry), review of medical history, review of medication history, Dallas pain questionnaire, and patient questionnaire (Oswestry Disability Index). Secondary efficacy outcome will assess subjective and objective parameters at three months, six months, and 12 months post CybroCell™ implantation and compare to those measured at baseline pre-implantation. These parameters will include visual analog scale, patient questionnaire (Oswestry Disability Index), Beck Depression Inventory, Dallas pain questionnaire, range of motion test, and radiological assessment using MRI to evaluate morphological changes to the treated discs.
We are currently in the process of finalizing the experimental cell bank production which will be transferred to a contract development and manufacturing organization, or CDMO, for the manufacturing of the master cell bank and working cell bank per FDA requirements and will submit the necessary documentation to the FDA. Our quotes from the CDMO for carrying out this work have been obtained.
We have completed two animal studies. Sixteen animals were used in the first pilot study (PMID 27853661)3 with the objective of determining the effects of intradiscal transplantation of neonatal human dermal fibroblasts, or nHDFs, on intravertebral disc, or IVD, degeneration by measuring disc height, magnetic resonance imaging, or MRI, signal intensity, gene expression, and collagen immunostaining. The results indicated that in the nHDF group there was a 10% increase in disk height index after eight weeks of treatment with a p value of <.05, while there was no significant difference in the saline treated group. When compared with the saline treated group, discs treated with nHDFs showed reduced expression of inflammatory markers, a higher ratio of collagen type II over collagen type I gene expression, and more intense immunohistochemical staining for both collagen types I and II. In the second study (PMID 30142460)4 38 animals were used with the objective of determining the impact of donor source on the therapeutic effect of dermal fibroblast treatment on disc degeneration and inflammation when comparing rabbit dermal fibroblasts, or RDFs, to nHDFs. Eight weeks after treatment, disc height indexes of discs treated with nHDF increased significantly by 7.8% (p<.01), whereas those treated with saline or RDF increased by 1.5% and 2.0%, respectively. Gene expression analysis showed that discs transplanted with nHDFs and RDFs displayed similar inflammatory responses (p=.2 to .8). Compared to intact discs, expression of both collagen types I and II increased significantly in nHDF-treated discs (p<.05), trending to significant in RDF-treated discs, and not significantly in saline treated discs. The ratio of collagen type II/collagen type I was higher in the IVDs treated with nHDFs (1.26) than those treated with RDFs (0.81) or saline (0.59) and intact discs (1.00). Last, proteoglycan contents increased significantly in discs treated with nHDF (p<.05) and were trending toward significance in the RDF- treated discs compared to those treated with saline. The results from the studies were positive and resulted in “first in human” trial approval. The technology allowed for differentiation of the HDFs into chondrocytes and the cells thrived in the spinal disc environment. The results showed the cells remained in the disc and did not migrate. Further, the cells created a biologic condition which appeared to increase the disc height.
3 Source: Chee A, Shi P, Cha T, Kao TH, Yang SH, Zhu J, Chen D, Zhang Y, An HS. Cell Therapy with Human Dermal Fibroblasts Enhances Intervertebral Disk Repair and Decreases Inflammation in the Rabbit Model. Global Spine J. 2016 Dec;6(8):771-779. doi: 10.1055/s-0036-1582391. Epub 2016 Apr 13. PMID: 27853661; PMCID: PMC5110358.
4 Source: Shi P, Chee A, Liu W, Chou PH, Zhu J, An HS. Therapeutic effects of cell therapy with neonatal human dermal fibroblasts and rabbit dermal fibroblasts on disc degeneration and inflammation. Spine J. 2019 Jan;19(1):171-181. doi: 10.1016/j.spinee.2018.08.005. Epub 2018 Aug 22. PMID: 30142460.
| 82 |
Below is a summary of animal study results from Howard An, M.D., Director, Spine Fellowship Program, Rush University Medical College:
Our studies have shown that this biological treatment using human dermal fibroblast cells, has great potential as a cell therapy for disc degeneration. When these cells were injected into a degenerating rabbit disc, they were retained in the disc for up to 8 weeks. Collagen Type II gene expression, a marker for disc repair and regeneration, was higher in the discs treated with human dermal fibroblast cells than those in the control treatment. Also higher in the cell treated discs were the disc heights and cell number. Together, this data suggests that human dermal fibroblast cells are a promising option for cell therapy to restore the biological function and reduce symptoms of intermediate or progressive degenerative discs.
We have received IND clearance from the FDA, conditional upon approval of our master cell bank, to run a Phase 1/2 study for patients suffering from degenerative disc disease and will be conducting this study within the United States. A timeline will be determined through discussions with the FDA.
Market Opportunity
Degenerative disc disease therapeutics represents an approximately $26.0 billion per year market. In addition to therapeutics, degenerative disc disease results in approximately 1.2 million orthopedic surgeries per year, at a cost of approximately $60,000 to $100,000 each, in the United States alone. CybroCell™ could replace therapeutics or defer many of these surgeries if it proves successful in regenerating disc cartilage and alleviating pain.
CYMS101 for Multiple Sclerosis
Multiple Sclerosis
Multiple sclerosis, or MS, has been characterized into four distinct clinical subtypes, differing in the age of onset, aggressiveness and progression of the disease, and frequency of relapses. Most MS cases (85%) follow a relapsing-remitting pattern, or RRMS, with an average relapse every 12 to 18 months in an untreated population, and short-term episodes of neurologic deficits that resolve completely or almost completely. MS relapse is commonly defined as new or worsening symptoms that last 24 hours in duration and occur in the absence of fever or infection. Other patients may transition to a more aggressive disease form known as secondary progressive MS, or will experience steadily progressive neurologic deterioration without relapses, known as primary progressive MS.
There is no primary indicator test for MS, but common testing for suspected MS involves MRI studies, evoked potentials testing, lumbar puncture/spinal tap, and other objective functional tests.
Once a diagnosis of MS has been determined, ongoing periodic disability measurement testing will occur as a standard clinical practice. The first Disability Status Scale was introduced by Kurtzke in 1955 and was later enhanced in 1983 into the Expanded Disability Status Scale, or EDSS. Over time, the EDSS has become the standard against which most MS clinical outcome measures are compared. Eight functional neurological systems are measured by the EDSS including vision, brainstem, pyramidal, cerebellar, sensory, bowel/bladder, mental/cerebral and ambulation (500m walk).
Other disability measurement tests include the Scripps Neurological Rating Scale, which is an overall neurological assessment; the Nine-Hole Pin Test, which measures arm function; and the Timed 25-Foot Walk Test, which measures ambulation function. The RAND 36-Question Health Survey may also be used, which is a general Quality of Life survey utilized by managed care organizations and by Medicare for routine monitoring and assessment of care outcomes in adult patients.
Available Treatments for MS
There is no known cure for MS. Treatments available for MS include steroids for temporary flare-ups, disease-modifying drugs, and drugs that target specific symptoms such as balance, vision, spasticity, sexual dysfunction, and bladder or bowel control. The mechanism of action of current MS disease-modifying drugs is to block the host’s immune-mediated attacks on the nerves to inhibit or minimize the progressive destruction of myelin. While these drugs may reduce the frequency of exacerbations and slow the disease progression from inducing further nerve damage, there is no myelin or nerve regenerative capability in any of them to restore the cumulative damage already in place. Additionally, as the disease progresses further, the ability for any of these drugs to effectively block immune-mediated myelin or nerve destruction becomes more blunted. Most MS drugs come with identified risks and side effects, including “black box” warnings.
| 83 |
Key companies providing existing MS treatments include:
| ● | Biogen, Inc.: Strong presence in the global market coupled with a diverse portfolio of MS drugs; | |
| ● | F. Hoffmann-La Roche Ltd (commonly known as Roche): Strong focus on research and development activities to develop novel medicine for MS treatment; and | |
| ● | Novartis AG: Increased focus on investment in research and development of innovative molecules. |
New Treatments Being Developed
Research and development in the neurological therapy area has always been active. Various novel molecules are being investigated for the treatment of MS. Some of the key pharmaceutical players are emphasizing the improvement of the disabilities associated with MS. The pipeline portfolios of various companies include agents with different mechanisms of action, which are expected to boost their demand from physicians, aiming to change the treatment algorithm in the coming years.
For a decade, various companies such as Sanofi, Johnson & Johnson, and Novartis AG, among others, have been investing in the treatment for MS to bring novel therapeutics with high efficacy and potency for patients. These companies have recently launched therapeutics intended for the most prevalent form of MS. In August 2020, the FDA approved Novartis AG’s Kesimpta, the only self-administered, targeted B-cell therapy for patients with relapsing MS, and in March 2021, Johnson & Johnson received FDA approval for the launch of Ponvory as a daily oral drug for treatment against MS.
The cost, outcome and quality of drugs approved is a priority of physicians as well as patients. Physicians play an important role in developing an interdisciplinary approach for the management of MS, which is a key cause for manufacturers to focus on novel molecules with different mechanisms of action. For instance, TG Therapeutics, Inc.’s ublituximab was recently approved by the FDA for the treatment of relapsing forms of MS, to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
The approval and commercialization of these recently approved drugs for the treatment of symptoms associated with MS is expected to boost the MS drug market growth globally in the near future.
Our Solution
We are developing CYMS101 as an allogeneic fibroblast cell-based therapy to treat MS. After completing animal studies using CYMS101 (allogeneic fibroblast cells), we received approval from Mexico for the conduct of clinical investigations using the fibroblast cell composition for patients with MS, and have completed the Phase 1 study called “Feasibility Study of Tolerogenic Fibroblasts in Patients with Refractory Multiple Sclerosis.” The study was conducted in five participants. The primary objective of the study was to assess safety, and the secondary objective was to assess efficacy. The results of the study for safety were no adverse effects during intravenous injection of the tolerogenic fibroblasts, no short or long-impact in complete blood count test during the 16-week monitoring period, and no short or long impact in electrocardiogram results during the 16-week monitoring period. In addition, the results of the study for efficacy included:
| ● | Paced Auditory Serial Addition Test (PASAT). The test was developed by Gronwell in 1977 and later adapted by Rao in 1989 for use in MS. The test is a measure of cognitive function that assesses auditory information processing speed as well as calculation ability. In the test, single digits are presented every 3 seconds and the patient must add each new digit to the one immediately prior to it. Scoring is the total number of correct responses out of 60 possible. There was a general improvement in PASAT score for all patients during the 16-week monitoring period. |
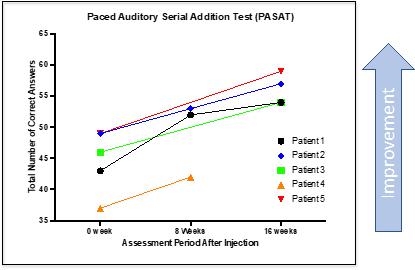
| 84 |
| ● | Nine-Hole Peg Test. This is a standardized quantitative test of upper extremity function. The test is the most frequently used measure of upper extremity function in MS. In the test, dominant and non-dominant hands are tested twice. In the test, pegs are picked up one at a time and placed in one of nine holes. Once all nine pegs are placed, the patient then removes the pegs one at a time and places them back in the container. Scoring is the time required to place pegs in holes and then remove the pegs from holes. General improvement of Nine-Hole Peg Test completion time for all patients during the 16-week testing period was noted. |
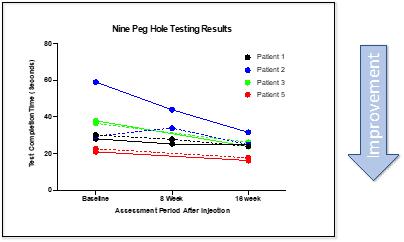
| ● | Timed 25-Foot Walk Test. The test is a quantitative mobility and leg function performance test. In the test, the patient is instructed to walk a 25 feet long path quickly but safely. The test is administered again by having the patient walk back the same distance. The score of the test is the average time of the two completed walks. No general improvement or deterioration was noted with the Timed 25-Foot Walk Test. |
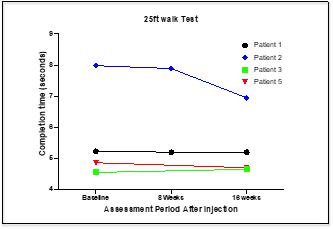
| ● | Expanded Disability Status Scale (EDSS). The test is used to quantify disability in MS and monitor changes in the level of disability over time. EDSS is widely used in clinical trials for assessment of participants with MS. The scale is based on neurological examination and impact on functional systems representing the network of neurons in the brain. EDSS scoring can vary widely due to complex scoring rules and subjective nature of the neurological testing. No general improvement or deterioration was noted with EDSS test, and no patient exhibited further deterioration during the study. |
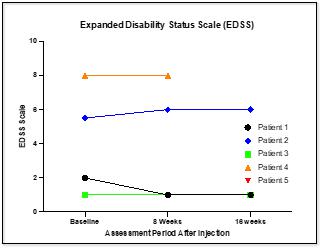
Although determining the mode of action is not necessary for filing for FDA approval (Brown and Wobst, 2020), and to date between 10-20% of approved drugs have no known mode of action determined (Moffat et al., 2017), we are currently conducting further research to determine the mode of action of fibroblasts in oligodendrocyte expansion. We will file an IND application for a Phase 2 clinical trial in MS in the United States. While determining the mode of action will be optimal, having a general sense of possible mode of action will have a tangible benefit in the development, optimization and mitigation of possible side-effects. We will likely seek a strategic partner to collaborate with us on the development of CYMS101 either before initiating the Phase 2 study, or after its completion, if successful, and prior to commencing with a Phase 3 clinical trial.
Market Opportunity
The MS drug market’s annual revenue is presently approximately $24.0 billion globally, with 48% of the revenues generated in the United States. The key companies in this market include Biogen Inc., F. Hoffmann-La Roche Ltd,, Sanofi, and Novartis AG. While there are more than 20 approved treatments, most of them have serious adverse effects and there are presently no cures. Both private and public organizations are increasing their investments in search of better treatments for this complex disease, including treatments that restore lost function, and government initiatives to improve access to MS drugs in developing economies are another driver of future growth in the MS market.
| 85 |
CYWC628 for Wound Healing
Wound Care/Healing
A chronic wound is one that is usually arrested in the inflammatory stage and cannot progress to the proliferative and remodeling phase of healing. Proinflammatory cytokines produced by necrotic tissue, foreign material and bacteria allow the inflammatory stage to continue. In addition, changes in the cellular deoxyribonucleic acid, or DNA, synthesis leads to increased formation of metalloproteases that impede the body’s attempt to heal by overwhelming the building blocks—chemotactant factors, growth factors and mitogens—needed for normal wound healing. Fibroblasts, an essential cell in the wound healing process, is epigenetically altered in the setting of chronic wounds so that their ability to replicate as well as produce the necessary building blocks for the formation of granulation tissue is altered. Further, the keratinocytes at the periphery of the wounds are phenotypically different so that while being able to proliferate, they cannot fully differentiate into migrating keratinocytes. This explains the epithelial build up often seen around the edge of the wound.
Diabetic foot ulcers are the most prominent type of chronic wounds. The rising prevalence of chronic diseases globally, is leading to increased incidence of chronic wounds, including diabetic foot ulcers, pressure ulcers and venous leg ulcers. These chronic wounds, especially late-stage “hard-to-heal ulcers,” exert a huge economic cost burden on healthcare agencies globally. Furthermore, over 50% of diabetic foot ulcers become infected, which raises the risk of hospitalization, amputation and death.
Available Wound Care Treatments
Several treatments are presently available for treatment of chronic wounds, including Apligraf, Grafix, DermACELL and TheraSkin. Apligraf is comprised of neonatal keratinocytes, and neonatal fibroblasts within a bovine collagen matrix, and may be used to treat venous leg ulcers and diabetic foot ulcers. Grafix is a cryopreserved human placental membrane that may be used as a wound cover, wrap and/or barrier to treat acute and chronic wounds, diabetic ulcers, pressure injuries, surgical wounds, burns and venous ulcers. DermACELL is a technologically advanced dermal matrix comprised of intact cellular matrix that has at least 97% of DNA removed and may be used in the treatment of chronic wounds such as diabetic foot ulcers. TheraSkin is a cryopreserved human skin allograft with both epidermis and dermis layers that may be used to promote wound healing.
New Treatments Being Developed
Increasing application of bioactive therapies like skin grafting and growth factors in urgent treatment of wounds like diabetic foot ulcers is resulting in high investment of market players in research and development of these therapies. Accordingly, the rise in adoption rate of advance bioactive therapies for rapid wound healing is expected to propel the growth of wound care market. For example, in February 2021, Axio Biosolutions Private Limited received CE mark from Europe for its MaxioCel advanced wound care product. The bioactive microfiber gelling technology helps wounds heal quickly.
Distinct clinical benefits offered by negative pressure wound therapy, or NPWT, coupled with introduction of new advanced features such as single use and portability, among others, for effective wound care is boosting the demand for NPWT devices from healthcare professionals globally. In January 2021, Smith & Nephew plc published that its PICO single-use negative pressure wound therapy system significantly reduced surgical site infections by 63.0% and the dehiscence by 30.0%. In January 2019, Applied Tissue Technologies LLC received FDA approval for its Platform Wound Dressing, NPWT device that eliminates the use of foam or gauze dressings. In April 2019, PolarityTE, Inc. launched clinical trials for its SkinTE regenerative tissue product for chronic wounds. The trials will evaluate SkinTE’s effectiveness in treating diabetic foot ulcers and venous leg ulcers.
Moreover, new players are entering the wound care market by focusing on allograft, xenograft, nanofibre, dermal substitutes and cell-based therapies to cater the unmet needs and growing demand for urgent and effective treatment among patients.
| 86 |
Our Solution
We are in the early stages of developing CYWC628 as an allogeneic fibroblast cell-based therapy for wound healing. Our studies are presently focused on utilizing fibroblasts and fibroblast-derived cells to treat wounds in diabetic mice and rats. Based upon our results achieved to date, we plan to pursue an IND submission with the FDA for wound healing as early as 2024.
Market Opportunity
The wound care market size was valued at approximately $17.0 billion globally in 2021, with more than half of the revenue generated in the United States and Europe, and, according to Fortune Business Insights published in March 2022, was projected to grow to approximately $28.0 billion by 2029. The rising prevalence of chronic diseases globally is leading to increased incidence of chronic wounds, including diabetic foot ulcers, pressure ulcers and venous leg ulcers. The huge economic cost burden exerted by chronic and acute wounds has led to an increase in initiatives being undertaken by governments globally, to create awareness among the general population for early diagnosis of wounds. These initiatives, along with improving reimbursement policies for wound care in these countries, are anticipated to drive the adoption of wound care products and lead to continued growth in this market.
Our Early-Stage Research
CYTER915 for Extension of Life
Extension of Life
Fibroblasts are no longer considered as mere structural components of organs but as dynamic participants in immune processes. Fibroblasts produce an environment that influences regulatory T cell migration, proliferation and activity, to ensure immunotolerance.
One of the key organs of the immune system is the thymus. It serves a vital role in T cell maturation and selection, elimination of self-reactive cells, establishment of central tolerance and T cell migration to recognize a wide range of pathogens. A variety of cells have been identified inside the thymus. These include epithelial cells, thymocytes, dendritic cells, or DC, macrophages, B lymphocytes, myoid cells, endothelial cells and fibroblasts. With age, the thymus declines in functionality through a process referred to as thymus or thymic involution. Publications have indicated that the process of involution enhances regulatory T cell generation which leads to increased susceptibility to pathogen infections, tumors and autoimmune diseases.
The thymus is critically important to the immune system, which serves as the body’s defense mechanism providing surveillance and protection against diverse pathogens, tumors, antigens and mediators of tissue damage. The immune system comprises a complex network of cellular and molecular components subdivided into thymus-independent (innate) and thymus-dependent (adaptive) arms which function synergistically in all immune responses. Innate immunity constitutes the first line of defense and is mediated by innate immune cells such as tissue macrophages, DC and granulocytes which elicit their effector function within minutes to hours following antigen exposure. Innate cells become activated via germ-line encoded pattern recognition receptors, including toll like receptors and nucleotide oligomerization domain-like receptors, which recognize invariant features of pathogens (pathogen-associate molecular patterns) and tissue damage.
Once activated, innate cells such as macrophages and neutrophils can effectively clear antigens via phagocytosis. Other types of innate cells, such as DC, take up and process antigens, resulting in expression of antigenic epitopes in conjunction with their major histocompatibility complex, or MHC, or human leukocyte antigen molecules. These DC can then serve as antigen-presenting cells for the priming of the adaptive immune system. In this way, the early innate response is coupled to, and facilitates, adaptive immunity.
The adaptive immune system consists of T and B lymphocytes which express specific antigen recognition receptors and develop highly specialized effector functions with the ability to form long-term immunological memory. Both B cells and T cells develop from bone marrow-derived progenitors; while mature B cells are exported to the periphery directly from the bone marrow, T cell development, maturation and export require critical differentiation steps to occur in the thymus. Thymus-dependent T cell differentiation processes include expression of an antigen-specific cell surface T cell receptor through recombination of germline-encoded gene segments, and thymic “education” involving negative selection of potentially self-reactive T cells and positive selection of T cells with the capacity to recognize antigens encountered in the periphery. These important thymic processes ensure that T cells can recognize antigens in the context of self-MHC, but do not elicit self-reactivity.
| 87 |
The spleen is one of the key secondary lymphoid organs responsible for the rapid response of the immune system to pathogens in the blood, and to maintain a long-term adaptive response to such pathogens. The spleen also serves as the key organ for iron metabolism and erythrocyte homeostasis. The organ also functions as a key storage site for platelets and leukocytes. A variety of cells have been identified in the spleen, including endothelia cells, mesothelial cells, reticular cells, erythrocytes, granulocytes, mononuclear cells, hemopoietic cells, macrophages, dendritic cells, plasma cells, CD4+ and CD8+ T cells, and migrating B cells. With age, the structure and function of the spleen changes, leading to decreased ability to respond positively to vaccination, increased susceptibility to viral and bacterial pathogen infections, and increased incidence of autoimmune disease. Accordingly, there is a need for improving and extending the productive life of the thymus and spleen through cell therapy, which could lead to an extension of human life by defeating the diseases that are allowed to proliferate during the declining process of these vital glands.
Our Solution
Our research program is in the early stages and is being designed to regenerate or reinvigorate production of the thymus and/or spleen. The regeneration comprises organogenesis and/or T cell development, wherein the tissue is differentiated and/or expansion of epithelial cells uses activated or inactivated fibroblasts. In addition to fibroblasts, we anticipate using other agents such as nucleic acids, cytokines, chemokines, transcription factors, epigenetic factors, growth factors, hormones or a combination thereof. The population of cells may be activated in vitro or ex vivo. The next step in developing fibroblasts for thymic or splenic involution reversal will be to design and conduct preclinical studies to demonstrate whether thymic or splenic involution reversal can be achieved in animal models.
Market Opportunity
The global anti-aging therapeutics market was estimated to have exceeded $500.0 million in 2021 and is expected to experience annual double-digit growth over the next ten years as aging populations and standards of living are increasing globally. The demand for effective regenerative medicine solutions for the aging population is higher than ever. As a result, anti-aging therapeutics are being developed by several companies using stem cells and regenerative medicine to identify, prevent, cure, and reverse age-related dysfunctions, illnesses, and diseases.
TCB190 for the Treatment of Certain Cancers
Our research on certain cancers is just beginning and further information about the opportunity will be released as it becomes available.
Manufacturing and Supply
We currently produce our cell therapy product candidates at our laboratory facility in Houston, Texas. We are in the process of contracting with a CDMO for the transfer of our experimental cell bank to produce our master cell bank, working cell bank and our fibroblast cell-based product candidate to enable clinical trials. If our product candidates receive marketing approval, we will evaluate the longer-term feasibility of building our own cGMP manufacturing facility or continuing to outsource production to a CDMO for clinical testing and commercial supply. We presently rely on third parties for certain portions of the cell therapy manufacturing process and will likely continue to do so in the future.
Intellectual Property
We were formed in April 2021 as a spinout from FibroGenesis. In connection with our formation, we issued the equivalent of 8,750,000 shares of Series A Preferred Stock to FibroGenesis in exchange for a patent assignment agreement, or the Patent Assignment Agreement, and an intellectual property cross-license agreement, or the Intellectual Property Cross-License Agreement. The Patent Assignment Agreement transfers all right, title and interest to certain patents/applications from FibroGenesis to us, and the Intellectual Property Cross-License Agreement allocates between FibroGenesis and us, exclusive fields of use for both assigned and retained patents issued/pending.
| 88 |
Through the Patent Assignment Agreement and the Intellectual Property Cross-License Agreement, FibroGenesis has effectively granted to us exclusive rights to develop fibroblasts in the following fields of use:
| ● | diagnosis, treatment, prevention and palliation of spinal diseases, disorders or conditions; | |
| ● | certain cancers; | |
| ● | orthopedic diseases, disorders or conditions; and | |
| ● | multiple sclerosis. |
FibroGenesis has retained exclusive rights for all other fields of use for both issued patents and patent applications transferred to us or retained by FibroGenesis.
The issued patents and patent applications assigned to us, along with additional patent applications filed independently by us after inception, include, as of the date hereof, a total of 48 patents and 109 patent applications pending. Our patent protections for our issued patents generally expire in years ranging from 2027 to 2043.
All of our issued patents are covered by the Patent Assignment Agreement and consist of 10 issued patents in the United States, eight issued patents in Australia, four issued patents in Japan, four issued patents in the United Kingdom, three issued patents in France, three issued patents in Germany, three issued patents in Italy, three issued patents in Spain, three issued patents in Hong Kong, two issued patents in Canada, two issued patents in China, two issued patents in Switzerland, and the remaining issued patent in Europe. One of our issued patents is also covered by the Intellectual Property Cross-License Agreement, which patent was issued in the United States.
Given present patent ineligibility laws concerning products of nature, there are presently no composition of matter patents covering CybroCell™, although there are patents related to the production of CybroCell™. We currently have patent applications pending for composition of matter for both CYMS101 and CYWC628.
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. We typically rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. We protect trade secrets and know-how by establishing confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and partners. These agreements generally provide that all confidential information developed or made known during the course of an individual or entity’s relationship with us must be kept confidential during and after the relationship. These agreements also generally provide that all inventions resulting from work performed for us or relating to our business and conceived or completed during the period of employment or assignment, as applicable, shall be our exclusive property. In addition, we take other appropriate precautions, such as physical and technological security measures, to guard against misappropriation of our property.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary and novel products and product candidates. Our competitors have developed, are developing or may develop products, product candidates and processes competitive with our product candidates. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. We believe that a significant number of products are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may attempt to develop product candidates. In addition, our products may need to compete with off-label drugs used by physicians to treat the indications for which we seek approval. This may make it difficult for us to replace existing therapies with our products.
We are developing CybroCellTM for the treatment of degenerative disc disease. Our competitors in the market for degenerative disc disease include Aesculap Implant Systems, LLC, Novartis AG, Pfizer Inc., Eli Lilly and Company, DiscGenics, Inc., Spine BioPharma, Inc. and Ferring B.V. In July 2021, Aesculap Implant Systems, LLC announced the long-term reporting from its pivotal trial for the activl® Artificial Disc.
We are developing CYMS101 as an allogeneic fibroblast cell-based therapy to treat MS. Key companies currently providing MS treatments include Biogen, Inc., F. Hoffmann-La Roche Ltd and Novartis AG. Various companies, such as Sanofi and Novartis AG, have been investing in the treatment for MS to bring novel therapeutics with high efficacy and potency for patients. These companies have recently launched therapeutics intended for the most prevalent form of MS. In August 2020, the FDA approved Novartis AG’s Kesimpta, the only self-administered, targeted B-cell therapy for patients with relapsing MS, and in March 2021, Johnson & Johnson received FDA approval for the launch of Ponvory as a daily oral drug for treatment against MS. TG Therapeutics, Inc.’s ublituximab for the indication of RMS was also recently approved by the FDA for treatment of MS.
We are in the early stages of developing CYWC628 as an allogeneic fibroblast cell-based therapy for wound healing. We face competition from several treatments presently available for treatment of chronic wounds, including Apligraf, Grafix, DermACELL and TheraSkin. In addition, increasing application of bioactive therapies like skin grafting and growth factors in urgent treatment of wounds like diabetic foot ulcers is resulting in high investment by companies in research and development of these therapies. In January 2019, Applied Tissue Technologies LLC received FDA approval for its Platform Wound Dressing, NPWT device that eliminates the use of foam or gauze dressings. In April 2019, PolarityTE, Inc. launched clinical trials for its SkinTE regenerative tissue product for chronic wounds. In January 2021, Smith & Nephew plc published that its PICO single-use negative pressure wound therapy system significantly reduced surgical site infections by 63.0% and the dehiscence by 30.0%, and in February 2021, Axio Biosolutions Private Limited received CE mark from Europe for its MaxioCel advanced wound care product, a bioactive microfiber gelling technology which helps wounds heal quickly.
| 89 |
Many of our current and potential competitors may have significantly greater financial, manufacturing, marketing, drug development, technical and human resources and commercial expertise than we do. Large pharmaceutical and biotechnology companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing biotechnology products. These companies also have significantly greater research and marketing capabilities than we do and may also have products that have been approved or are in late stages of development, and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical and biotechnology companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies, as well as in acquiring technologies complementary to, or necessary for, our programs. As a result, our competitors may succeed in obtaining approval from the FDA, the EMA or other comparable foreign regulatory authorities or in discovering, developing and commercializing products in our field before we do. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe effects, are more convenient, have a broader label, are marketed more effectively, are reimbursed or are less expensive than any products that we may develop. Technological advances or products developed by our competitors may render our technologies or product candidates obsolete, less competitive or not economical. If we are unable to compete effectively, our opportunity to generate revenue from the sale of any products we may develop, if approved, could be adversely affected.
Regulatory Environment
Government Regulation and Product Approval
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries and jurisdictions impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of products such as those we are developing. These entities regulate, among other things, the research, development, testing, manufacture, quality control, packaging, safety, effectiveness, labeling, storage, record keeping, approval, advertising, promotion, distribution, post-approval monitoring and reporting, sampling, export and import of our product candidates. Any product candidates that we develop must be approved by the FDA before they may be legally marketed in the United States and by the appropriate foreign regulatory agency before they may be legally marketed in those foreign countries. Generally, our activities in other countries will be subject to regulation that is similar in nature and scope as that imposed in the United States, although there can be important differences.
U.S. Product Development Process
In the United States, the FDA regulates drugs under the U.S. Federal Food, Drug, and Cosmetic Act, or the FDCA, and biologics under the FDCA and the Public Health Service Act and their implementing regulations. The processes for obtaining regulatory approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources. The failure to comply with applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant and/or sponsor to a variety of administrative or judicial sanctions, including refusal by the FDA to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters and other types of letters, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties brought by the FDA and the U.S. Department of Justice or other governmental entities. In addition, an applicant may need to recall a product. Additionally, certain of our product candidates are subject to regulation in the United States as a combination product. If marketed individually, each component would be subject to different regulatory pathways and would require approval of independent marketing applications by the FDA. A combination product, however, is assigned to a center within the FDA that will have primary jurisdiction over its regulation based on a determination of the combination product’s primary mode of action, which is the single mode of action that provides the most important therapeutic action. In the case of our CybroCell™ product candidate, we believe that the primary mode of action is attributable to the biologic component of the product. We expect to seek approval of this combination product candidate through a BLA, and we do not expect that the FDA will require a separate marketing authorization for each of the drug and biologic constituents of the product.
| 90 |
The process required by the FDA before a new product may be marketed in the United States generally involves the following:
| ● | completion of nonclinical or preclinical laboratory tests, animal studies and formulation studies in accordance with the FDA’s good laboratory practice, or GLP, requirements and other applicable regulations; |
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| ● | approval by an IRB or ethics committee at each clinical site before each trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials in accordance with GCP requirements to establish the safety and efficacy of the proposed drug for its intended use, or with respect to biologics, the safety, purity and potency of the product candidate for each proposed indication; |
| ● | submission to the FDA of an NDA or BLA after completion of all pivotal trials; |
| ● | a determination by the FDA within 60 days of its receipt of an NDA or BLA to file the application for review; |
| ● | satisfactory completion of an FDA advisory committee review, if applicable; |
| ● | satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity, and of selected clinical investigation sites to assess compliance with GCPs; |
| ● | a potential FDA audit of the preclinical and/or clinical trial sites that generated the data in support of the NDA or BLA; and |
| ● | the FDA’s review and approval of the NDA or BLA to permit commercial marketing of the product for particular indications for use in the United States. |
Preclinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as in vitro and animal studies to assess potential safety and efficacy. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations for safety/toxicology studies.
Prior to beginning the first clinical trial with a product candidate in the United States, we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical trials. Some preclinical studies may continue even after the IND is submitted. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
| 91 |
In addition to the submission of an IND to the FDA before initiation of a clinical trial in the United States, certain human clinical trials involving recombinant or synthetic nucleic acid molecules are subject to oversight at the local level as set forth in the NIH Guidelines. Specifically, under the NIH Guidelines, supervision of human gene transfer trials includes evaluation and assessment by an Institutional Biosafety Committee, or IBC, a local institutional committee that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment, and such review may result in some delay before initiation of a clinical trial. While the NIH Guidelines are not mandatory unless the research in question is being conducted at or sponsored by institutions receiving NIH funding of recombinant or synthetic nucleic acid molecule research, many companies, and other institutions not otherwise subject to the NIH Guidelines voluntarily follow them.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects, or their legal representative, provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the inclusion and exclusion criteria, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an independent IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site and must monitor the study until completed. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some clinical trials also include oversight by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee, which provides authorization for whether or not a trial may move forward at designated check points based on access to certain data from the trial and may recommend that the clinical trial be halted if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical trials and clinical trial results to public registries.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1: The product candidate is initially introduced into healthy human subjects or, in certain cases such as certain cancers, patients with the target disease or condition. These trials are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. In the case of some products for severe or life-threatening diseases, such as certain cancers, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| ● | Phase 2: The product candidate is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages, dose tolerance and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
| ● | Phase 3: The product candidate is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for physician labelling. Generally, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA or BLA. |
Post-marketing studies, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, such as with accelerated approval drugs, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval.
| 92 |
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2 clinical trials or before an NDA or BLA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach agreement on the next phase of development. Sponsors typically use the meetings at the end of a Phase 2 clinical trial to discuss the clinical trial’s results and present plans for a pivotal Phase 3 clinical trial that they believe will support approval of their new drug.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. In addition, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
While the IND is active and before product approval, progress reports summarizing the results of the clinical trials and nonclinical studies performed since the last progress report must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the same or similar drugs, findings from animal or in vitro testing suggesting a significant risk to humans, and any clinically important increased incidence of a serious suspected adverse reaction compared to that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information.
U.S. Review and Approval Process
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, preclinical and other nonclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling and other relevant information are submitted to the FDA as part of an NDA or BLA requesting approval to market the product. Data may come from company-sponsored clinical trials intended to test the safety and effectiveness of the use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational drug product to the satisfaction of the FDA. The submission of an NDA or BLA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances. Additionally, no user fees are assessed on NDAs or BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA reviews an NDA or BLA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant to assure and preserve the product’s identity, strength, quality and purity. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a new molecular entity or BLA to review and act on the submission. This review typically takes twelve months from the date the NDA or BLA is submitted to the FDA because the FDA has approximately two months to make a “filing” decision after the application is submitted. The FDA conducts a preliminary review of all NDAs or BLAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA or BLA for filing. In this event, the NDA or BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
| 93 |
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA or BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCPs. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
After the FDA evaluates an NDA or BLA, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete, and the application will not be approved in its present form. A Complete Response Letter usually describes the specific deficiencies in the NDA or BLA identified by the FDA and may require additional clinical data, such as an additional pivotal Phase 3 clinical trial or other significant and time-consuming requirements related to clinical trials, nonclinical studies or manufacturing. If a Complete Response Letter is issued, the sponsor must resubmit the NDA or BLA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the NDA or BLA does not satisfy the criteria for approval.
If regulatory approval of a product is granted, such approval will be granted for particular indications and may contain limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the NDA or BLA with a REMS to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy to manage a known or potential serious risk associated with a medicine and to enable patients to have continued access to such medicines by managing their safe use, and could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries, and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace. The FDA may also require one or more post-marketing studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization, and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could impact the timeline for regulatory approval or otherwise impact ongoing development programs.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the United States or, if it affects more than 200,000 individuals in the United States, there is no reasonable expectation that the cost of developing and making a drug product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan designation must be requested before submitting an NDA or BLA. After the FDA grants orphan designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan designation does not convey any advantage in or shorten the duration of the regulatory review and approval process. Orphan designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers.
| 94 |
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Competitors, however, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity. Orphan exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same drug as defined by the FDA or if our product candidate is determined to be contained within the competitor’s product for the same indication or disease. If an orphan designated product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan exclusivity. In addition, exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Expedited Development and Review Programs
The FDA has a number of programs intended to expedite the development or review of products that meet certain criteria. For example, new drugs are eligible for Fast Track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a fast track product has opportunities for more frequent interactions with the review team during product development, and the FDA may consider for review sections of the NDA or BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA or BLA, the FDA agrees to accept sections of the NDA or BLA and determines that the schedule is acceptable, and the sponsor pays any required user fee upon submission of the first section of the NDA or BLA.
A product, including a product with a Fast Track designation, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. A product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug designated for priority review in an effort to facilitate the review. The FDA endeavors to review applications with priority review designations within six months of the filing date as compared to ten months for review of standard NDAs or BLAs under its current PDUFA review goals.
In addition, a product may be eligible for accelerated approval. Products intended to treat serious or life-threatening diseases or conditions may be eligible for accelerated approval upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. In addition, the FDA currently requires pre-approval of promotional materials as a condition for accelerated approval, which could adversely impact the timing of the commercial launch of the product.
A product candidate intended to treat a serious or life-threatening disease or condition may also be eligible for breakthrough therapy designation to expedite its development and review. A product can receive breakthrough therapy designation if preliminary clinical evidence indicates that the product, alone or in combination with one or more other drugs or biologics, may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the fast track program features, as well as more intensive FDA interaction and guidance beginning as early as Phase 1 and an organizational commitment to expedite the development and review of the product candidate, including involvement of senior managers.
| 95 |
In addition, the FDA may designate a product as a regenerative medicine advanced therapy, or RMAT. The RMAT designation is intended to facilitate an efficient development program for, and expedited review of, any product candidate that meets the following criteria: (i) the product candidate qualifies as a RMAT, which is defined as a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, with limited exceptions; (ii) the product candidate is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and (iii) preliminary clinical evidence indicates that the product candidate has the potential to address unmet medical needs for such a disease or condition. RMAT designation provides potential benefits that include more frequent meetings with the FDA to discuss the development plan for the product candidate, and eligibility for rolling review and priority review of BLAs. Cell therapy candidates granted RMAT designation may also be eligible for accelerated approval on the basis of a surrogate or intermediate endpoint reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of sites, including through expansion to additional sites, as appropriate. RMAT-designated cell therapy candidates that receive accelerated approval may, as appropriate, fulfill their post-approval requirements through the completion of clinical trials, patient registries, or through submission of other sources of real world evidence, such as electronic health records, through the collection of larger confirmatory data sets, or via post-approval monitoring of all patients treated with such therapy prior to approval of the therapy.
Fast track designation, breakthrough therapy designation, priority review, RMAT designation and accelerated approval do not change the standards for approval, but may expedite the development or approval process. Even if a product candidate qualifies for one or more of these programs, the FDA may later decide that the product candidate no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
Post-Approval Requirements
Any products manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, product sampling and distribution, and advertising and promotion of the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing user fee requirements, under which the FDA assesses an annual program fee for each product identified in an approved NDA or BLA. Drug and biologic manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMPs, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMPs and impose reporting requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMPs and other aspects of regulatory compliance.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program.
Other potential consequences include, among other things:
| ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● | fines, warning letters, or untitled letters; |
| ● | clinical holds on clinical trials; |
| ● | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; |
| ● | consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs; |
| ● | mandated modification of promotional materials and labeling and the issuance of corrective information; |
| ● | the issuance of safety alerts, Dear Healthcare Provider letters, press releases and other communications containing warnings or other safety information about the product; or |
| ● | injunctions or the imposition of civil or criminal penalties. |
| 96 |
The FDA also may require post-marketing testing, known as Phase 4 testing, and surveillance to monitor the effects of an approved product. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures.
The FDA closely regulates the marketing, labeling, advertising and promotion of drug products. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe, in their independent professional medical judgment, legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products. The federal government has levied large civil and criminal fines against companies for alleged improper promotion of off-label use and has enjoined companies from engaging in off-label promotion. The FDA and other regulatory agencies have also required that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. However, companies may share truthful and not misleading information that is otherwise consistent with a product’s FDA-approved labeling.
In addition, the distribution of prescription biopharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription biopharmaceutical product samples and impose requirements to ensure accountability in distribution.
Biosimilars and Reference Product Exclusivity
The Biologics Price Competition and Innovation Act of 2009, or BPCIA, created an abbreviated approval pathway for biological products that are highly similar, or “biosimilar,” to or interchangeable with an FDA-approved reference biological product. The FDA has issued several guidance documents outlining an approach to review and approval of biosimilars. Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, is generally shown through analytical studies, animal studies, and a clinical trial or trials. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. A product shown to be biosimilar or interchangeable with an FDA-approved reference biological product may rely in part on the FDA’s previous determination of safety and effectiveness for the reference product for approval, which can potentially reduce the cost and time required to obtain approval to market the product.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of its product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law.
A biological product can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
| 97 |
Data Privacy and Security
Other federal legislation may affect our ability to obtain certain health information in conjunction with our research activities. We may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. The Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and its implementing regulations, collectively referred to as HIPAA, imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information. HIPAA also prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statements or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services. We may obtain health information from third parties, such as research institutions, that are subject to privacy and security requirements under HIPAA. Although we are not directly subject to HIPAA, other than with respect to providing certain employee benefits, we could potentially be subject to criminal penalties if we, our affiliates, or our agents knowingly obtain or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA.
In addition, numerous federal and state laws and regulations that address privacy and data security, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), govern the collection, use, disclosure and protection of health-related and other personal information. Failure to comply with data protection laws and regulations could result in government enforcement actions and create liability for us, which could include civil and/or criminal penalties, private litigation and/or adverse publicity that could negatively affect our business.
Failure to achieve and sustain compliance with applicable federal and state privacy, security and fraud laws could result in government enforcement actions and create liability for us, which could include civil and/or criminal penalties, private litigation and/or adverse publicity that could negatively affect our operating results and business.
Other U.S. Regulatory Requirements
Biopharmaceutical companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business that may constrain the financial arrangements and relationships through which we research, as well as sell, market and distribute any products for which we obtain marketing authorization. Such laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, and transparency laws and regulations related to drug pricing and payments and other transfers of value made to physicians and other healthcare providers. If their operations are found to be in violation of any of such laws or any other governmental regulations that apply, they may be subject to penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, the curtailment or restructuring of operations, integrity oversight and reporting obligations, exclusion from participation in federal and state healthcare programs and responsible individuals may be subject to imprisonment.
| 98 |
Coverage and Reimbursement
Sales of any product depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state, and foreign government healthcare programs, commercial insurance and managed healthcare organizations, and the level of reimbursement for such product by third-party payors. In the United States, no uniform policy of coverage and reimbursement for products exists among third-party payors and coverage and reimbursement levels for products can differ significantly from payor to payor. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. Factors payors consider in determining reimbursement are based on whether the product is (i) a covered benefit under its health plan, (ii) safe, effective and medically necessary, (iii) appropriate for the specific patient, (iv) cost-effective and (v) neither experimental nor investigational. Decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. These third-party payors are increasingly reducing reimbursements for medical products, drugs and services. For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult because of the higher prices often associated with such drugs.
In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit sales of any product. Decreases in third-party reimbursement for any product or a decision by a third-party payor not to cover a product could reduce physician usage and patient demand for the product and also have a material adverse effect on sales.
Healthcare Reform
In March 2010, the ACA was enacted, which substantially changed the way healthcare is financed by both governmental and private insurers, and significantly affected the biopharmaceutical industry. The ACA contained a number of provisions, including those governing enrollment in federal healthcare programs, reimbursement adjustments and changes to fraud and abuse laws. Additionally, the ACA:
| ● | increased the minimum level of Medicaid rebates payable by manufacturers of brand name drugs from 15.1% to 23.1% of the average manufacturer price; |
| ● | required collection of rebates for drugs paid by Medicaid managed care organizations; |
| ● | required manufacturers to participate in a coverage gap discount program, under which they must agree to offer 50% (increased to 70% pursuant to the Bipartisan Budget Act of 2018, effective as of January 1, 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and |
| ● | imposed a non-deductible annual fee on pharmaceutical manufacturers or importers who sell “branded prescription drugs” to specified federal government programs. |
Other legislative changes have been proposed and adopted since the ACA was enacted, including aggregate reductions of Medicare payments to providers of 2% per fiscal year. Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries, proposed and enacted legislation and executive orders issued by the former Trump administration designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. Individual states in the United States have also become increasingly active in implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
International Regulation
In order to market any product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of products. Whether or not it obtains FDA approval for a product, an applicant will need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can commence clinical trials or marketing of the product in those countries or jurisdictions.
| 99 |
The regulation of our product candidates outside of the United States varies by country. Certain countries regulate human tissue products as a pharmaceutical product, which would require us to make extensive filings and obtain regulatory approvals before selling our product candidates. Certain other countries classify our product candidates as human tissue for transplantation but may restrict its import or sale. Other countries may have no application regulations regarding the import or sale of products similar to our product candidates, creating uncertainty as to what standards we may be required to meet.
Employees
As of September 30, 2023, we had eight full-time employees, including five employees with medical or doctoral degrees and six employees directly engaged in research and development, with the rest providing administrative, business and operations support. None of our employees are represented by labor unions or covered by collective bargaining agreements. We consider the relationship with our employees to be good.
Our Facilities
Our principal executive offices are located at 455 E. Medical Center Blvd., Suite 300, Houston, Texas, where we lease approximately 23,000 square feet of office space. The space serves as the location of our corporate headquarters. The lease expires in April 2027. In addition, we have leased research labs and offices in Houston, Texas, for our research and cell manufacturing operations.
We believe that our facilities are adequate for our current and anticipated near-term needs and that suitable additional or substitute space would be available if needed.
Legal Proceedings
From time to time, we may be party to litigation arising in the ordinary course of business. We are currently not a party to any material legal proceedings and, to the best of our knowledge, no material legal proceedings are currently pending or threatened. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
| 100 |
Executive Officers
The
following table sets forth certain information, as of the date of this prospectus, concerning our executive officers:
| Name | Age | Position | ||
| Pete O’Heeron, MSHA | 60 | Founder, Chairperson and Chief Executive Officer | ||
| Mark Andersen, CPA CFA | 52 | Chief Financial Officer | ||
| Hamid Khoja, Ph.D. | 54 | Chief Scientific Officer |
The following is a biographical summary of the experience of our executive officers.
Pete O’Heeron, MSHA. Pete O’Heeron founded our company and has served as our Chief Executive Officer, and the Chairperson and member of our board of directors since our inception in April 2021. Mr. O’Heeron is also the founder of FibroGenesis, our affiliate, and has served as the Chief Executive Officer of FibroGenesis since January 2006. Mr. O’Heeron is a preeminent biopharma inventor, with over 300 patents issued and pending in the areas of biologics, cell therapy and medical devices. Mr. O’Heeron is a seasoned leader in his field, with over 25 years of experience in medical technology and biotech development. As Chief Executive Officer, he aims to position us to become a global leader in fibroblast-based cell therapies with the development and commercialization of therapies that can cure and treat patients suffering from chronic diseases. Mr. O’Heeron brings together multi-disciplinary teams and resources necessary to commercialize unique technologies. Prior to founding our company and FibroGenesis, he founded an operational investment group, Advanced Medical Technologies, LLC, that identified early-stage opportunities in the medical field with strong intellectual property potential in 2006. He also founded in 1998 NeoSurg Technologies, which developed the T2000 Minimally Invasive Access System. NeoSurg Technologies was sold to Cooper Surgical in 2006. Mr. O’Heeron also previously served in a variety of executive-level positions at Christus Health Care Corporation from 1988 until 1995 and has provided strategic advisory services to healthcare companies in the areas of biologics, advanced surgical instrumentation and telemedicine. Mr. O’Heeron received his Bachelor’s Degree in Healthcare Administration from Texas State University, his Masters in Healthcare Administration from the University of Houston Clear Lake, and his Executive Management Certification in Mergers and Acquisition from the University of Chicago. We believe Mr. O’Heeron is qualified to serve as a member of our board of directors based on our review of his experience, qualifications, attributes and skills, including co-founding our company and his executive leadership experience in the biotechnology industry.
Mark Andersen, CPA CFA. Mark Andersen has served as our Chief Financial Officer since June 2022. Prior to joining us, Mr. Andersen most recently served as Chief Financial Officer and Vice President of Administration for the Indiana Biosciences Research Institute in Indianapolis, Indiana, from May 2016 until May 2022. In that role, he was responsible for finance, human resources, legal, and information technology for the institute. Mr. Andersen helped create the operating infrastructure for the institute, assisted with fundraising and provided oversight for the endowment investment portfolio, which grew to nearly $150.0 million. Prior to that, from August 2015 until February 2016, Mr. Andersen served as Vice President Finance and Corporate Controller for MiMedx with responsibility for SEC reporting and finance functions. Previously, from January 2004 to August 2015, Mr. Andersen held multiple financial leadership roles at Eli Lilly and Company, including Investments Director for the company’s pension plan, Finance Director for Mergers and Acquisitions, and Controller for Lilly USA. Mr. Andersen received his Bachelor of Science degree in accounting and Master of Science in accountancy from Southern Utah University, and his MBA from the University of Michigan Ross School of Business.
| 101 |
Hamid Khoja, Ph.D. Hamid Khoja has served as our Chief Scientific Officer since August 2021. Dr. Khoja has more than 25 years of experience as a leader of scientific teams, development of cell-based genomic, proteomic, epigenetics assays, and tools, protocols and technologies for use in drug discovery and development and clinical diagnostics. Prior to joining us, Dr. Khoja most recently served from March 2009 to August 2021 as the Principal Scientist as Covaris, LLC, a privately-held scientific tools company with emphasis in genomics, epigenetics, and proteomics, where he provided long-term strategic applications proposals to the Chief Executive Officer, managed external collaborations for product and applications development, assessed new technologies for acquisition and OEM opportunities, and presented posters and presentations at numerous scientific conferences. Dr. Khoja led the effort in successfully incorporating Covaris technology into the Illumina Next Generation Sequencing technology protocols leading to over 15,000 citations. Dr. Khoja also developed the Covaris chromatin immunoprecipitation methodology with over 3,000 citations in peer-reviewed publications, as well as leading the effort in using Covaris technology for simplifying epigenetics assay workflows for use in drug development and discovery, and clinical use. Dr. Khoja also led collaborations with the U.S. National Cancer Institute for successful development of microbiome DNA extraction using acoustics, and completion of FDA EUA SARA-CoC-2 bridge study design for approval of new sample collection and viral ribonucleic acid (RNA) extraction using Covaris technology. Dr. Khoja also developed a patented workflow for the manufacturing of synthetic cell-free DNA for use as reference standard in sequencing based liquid biopsy clinical oncology-based assays. Prior to Covaris, Dr. Khoja was a Senior Applications Scientist at Genomic Solutions, a startup scientific tools company later acquired by Harvard Apparatus, from March 2022 to March 2009, where he led the development of a high throughput protein crystallization platform used in pharmaceutical industry for drug development, managed the scientific applications group, presented company resources at scientific meetings and assessed new technologies for acquisition and OEM opportunities. During the startup phase of Sequenom, Inc., from January 2000 to March 2003, Dr. Khoja established the methodology for highly multiplexed polymerase chain reaction, or PCR, used in the development of Sequenom’s massEXTEND technology for MALDI-TOF MS-based analysis of single nucleotide polymorphisms and genetic disease. Dr. Khoja led the effort in developing diagnostic MS-based assays for hemochromatosis, cystic fibrosis and ten predominantly Jewish genetic diseases using Sequenom’s massEXTEND technology which were then transferred to a large clinical diagnostic company. Dr. Khoja also previously worked at Eli Lilly and Company from November 1998 to September 1999 and Chiron Corporation from October 1995 to October 1998. During his career at Eli Lilly, Dr. Khoja established a high throughput PCR and sequencing strategy using a variety of sequencing strategies and bioinformatic tools available in 1999 for obtaining high coverage genome sequencing which led to the finalizing of the first ever complete sequence of the S. pneumoniae genome. At Chiron Corporation, which was subsequently acquired by Novartis, Dr. Khoja helped in the design, development and optimization of HTP binding assays for FGFR, VEGF, PDGF, and EPO receptors, identification of novel g-protein coupled seven transmembrane receptors, and identification of novel proteins involved in the TNF signaling pathway, and development of branched-DNA based HTP screening for ligand-induced oncogene quantification.
Dr. Khoja received his Bachelor of Science in Molecular Biology from the University of Southern California and his Ph.D. in Molecular Biology from Boston University.
Non-Employee Directors
The following table sets forth certain information, as of the date of this prospectus, concerning our non-employees who serve on our board of directors:
| Name | Age | Position | ||
| Robert Hoffman, CPA (inactive) | 57 | Director | ||
| Victoria Niklas, M.D. | 64 | Director | ||
| Richard Cilento, Jr., MBA | 61 | Director | ||
| Stacy Coen, MBA | 52 | Director | ||
| Matthew Link | 48 | Director |
The following is a biographical summary of the experience of our non-employee directors.
Robert Hoffman, CPA (inactive). Robert Hoffman has served on our board of directors since April 2021. Mr. Hoffman currently serves as President, Chief Executive Officer and Chairperson of the board of directors of Kintara Therapeutics, Inc. (Nasdaq: KTRA), a clinical stage, biopharmaceutical company focused on the development and commercialization of new cancer therapies, a member of the board of directors of ASLAN Pharmaceuticals Limited (Nasdaq: ASLN), an oncology-focused biotechnology company developing a portfolio of immuno-oncology agents and targeted therapies, and Chairperson, and a member, of the board of directors of Antibe Therapeutics Inc., a Toronto, Canada-based pharmaceutical company listed on the Toronto Stock Exchange. Mr. Hoffman previously served as Senior Vice President and Chief Financial Officer of Heron Therapeutics, Inc., (Nasdaq: HRTX), a commercial-stage biotechnology company, from April 2017 to October 2020, and as Chief Financial Officer of AnaptysBio, Inc. (Nasdaq: ANAB), a specialty pharmaceutical company, from July 2015 to September 2016. From June 2012 to July 2015, Mr. Hoffman served as the Senior Vice President, Finance and Chief Financial Officer of Arena Pharmaceuticals, Inc., or Arena, a biopharmaceutical company, prior to its acquisition by Pfizer Inc. in March 2022. From August 2011 to June 2012 and previously from December 2005 to March 2011, Mr. Hoffman served as Arena’s Vice President, Finance and Chief Financial Officer and in a number of various roles of increasing responsibility from 1997 to December 2005. Mr. Hoffman formerly served as a member of the board of directors of Saniona AB, a biopharmaceutical company, from September 2021 to May 2022, and as a member of the board of directors of Kura Oncology, Inc. (Nasdaq: KURA), a cancer research company, from March 2015 to August 2021. He also previously served as a member of the board of directors of CombiMatrix Corporation, a molecular diagnostics company, MabVax Therapeutics Holdings, Inc., a biopharmaceutical company, and Aravive, Inc. (Nasdaq: ARAV), a clinical stage biotechnology company. Mr. Hoffman serves as a member of the steering committee of the Association of Bioscience Financial Officers. Mr. Hoffman formerly served as a director and President of the San Diego Chapter of Financial Executives International and was an advisor to the Financial Accounting Standard Board, or FASB, from 2010 to 2020, advising the U.S. accounting rulemaking organization on emerging issues and new financial guidance. Mr. Hoffman holds a B.B.A. from St. Bonaventure University. We believe Mr. Hoffman’s financial and executive business experience qualifies him to serve on our board of directors.
| 102 |
Victoria Niklas, M.D. Victoria Niklas has served on our board of directors since April 2021. Dr. Niklas has a distinguished career spanning more than two decades in translational research, clinical care and teaching at academic health centers, and is currently the Chief Medical Officer of Oak Hill Bio, a clinical-stage neonatology and rare disease therapeutics company, a position she has held since 2022. Prior to joining Oak Hill Bio, Dr. Niklas served in Global Medical Affairs and as Global Program Leader of the OHB-607 program in Rare Disease and Hematology at Takeda Pharmaceuticals. Before Takeda, she was Chief Medical and Scientific Officer at Prolacta Bioscience, a neonatal nutritional product development company based on human donor milk. Dr. Niklas has over 20 years of experience as an academic neonatologist with expertise in developmental and acquired inflammatory disorders of the gut, the lung and the mucosal immune system with relevance to diseases across the lifespan. She has held positions as Chief, Division of Newborn Medicine at Nemours Children’s Hospital, Chief of Neonatology at UCLA Olive View Medical Center, and Visiting Professor of Clinical Pediatrics at the David Geffen School of Medicine at UCLA. Dr. Niklas is board certified in Perinatal and Neonatal Medicine and holds a California medical license. In addition to being a co-author on numerous scientific and clinical publications, she has helped lead the development of patented products and has served as a board member for multiple biotech and early-stage companies in functional foods. Dr. Niklas received her MD from Harvard Medical School, her MA in Biochemistry and Molecular Biology from Harvard University, and her bachelor’s in Biological Sciences from Goucher College. We believe Dr. Niklas’ extensive experience and knowledge in the biotechnology sector qualifies her to serve on our board of directors.
Richard Cilento, Jr., MBA. Richard Cilento has served on our board of directors since April 2021. Mr. Cilento is the founder, Chairperson of the board of directors and Chief Executive Officer of GlycosBio Inc., a life sciences research and development company. Mr. Cilento was the founder, President and Chief Executive Officer of FuelQuest, Inc., a provider of information technology, supply chain management and tax automation technologies, which was acquired by Saracen Energy Advisors LP in May 2007. Mr. Cilento has held senior-management positions with several technology firms, including Xerox Corporation, where he served as Vice President of Strategic Services of Xerox Connect. Prior to that, he was the Vice President of Corporate Services for XLConnect Solutions, where he served as the lead technologist for advanced systems and supported the organization through its initial public offering and its eventual merger with Xerox. An aeronautical and astronomical engineer, Mr. Cilento began his career at the U.S. National Aeronautics and Space Administration (NASA), where he and his team built space shuttle flight plans for the U.S. Department of Defense Star Wars program and a diverse set of government-funded technology and life science experimentation. Mr. Cilento was a lead engineer who designed and planned the space station assembly sequences for the construction of the International Space Station. Mr. Cilento holds a BS degree in Aeronautical and Astronomical Engineering from the University of Illinois and an MBA at the University of Houston. We believe Mr. Cilento’s business experience across a broad set of technical industries and executive-level knowledge of capital markets, including venture capital, private equity and public markets, qualifies him to serve on our board of directors.
Stacy Coen, MBA. Stacy Coen has served as a member of our board of directors since July 2021. Ms. Coen has over 25 years of business and corporate development experience from leading oncology and rare disease companies. She is currently the Chief Business Officer for ImmunoGen, Inc., a company that is developing the next generation of antibody-drug conjugates to improve outcomes for cancer patients. Prior to ImmunoGen, Ms. Coen worked at Editas Medicine, Inc., a biotechnology company developing therapies for rare diseases, where she served as Vice President, Business Development and was responsible for business development, strategy, transactions and alliance management. Prior to joining Editas, Ms. Coen served in multiple roles of increasing responsibility at Genzyme Corporation (now known as Sanofi Genzyme), including as Vice President, Head of Rare Disease Business Development and Licensing, and as Vice President, Global Head of Strategy and Business Development, Multiple Sclerosis, among others. Ms. Coen currently serves on the Huntington’s Disease Society of America’s Center Programs & Education Advisory Committee and is a member of MassBio and the Alliance for Regenerative Medicine. Ms. Coen received a BS in Finance and Economics from the University of Massachusetts and an MBA from the Darden Graduate School of Business at the University of Virginia. We believe Ms. Coen’s extensive executive-level experience in the biotechnology industry qualifies her to serve on our board of directors.
| 103 |
Matthew Link. Matthew Link has served on our board of directors since April 2021. Mr. Link has more than 20 years of experience in the healthcare and medical technology industries and currently serves as Chief Commercial Officer for Sight Sciences (SGHT). From 2021 to 2023 he served as managing partner at Orion Healthcare Advisors, LLC, a consulting services provider. From 2006 to 2021 Mr. Link served in regional and executive leadership positions at NuVasive Inc., a global leader in surgical implants and enabling technology for spine surgery and orthopedics. As President of NuVasive, Inc., his responsibilities included oversight of global business units in spine, neurophysiology, and orthopedics. Prior to NuVasive, Inc., Mr. Link held commercial leadership roles at Depuy Orthopedics and Depuy Spine. He also currently serves as chairman of the board of directors at Galen Robotics and as a member of the board of directors of Springbok Analytics and DinamicOR, and the Coulter Translational Research Endowment at the University of Virginia. Mr. Link received a BSEd in Physical Education and Sports Medicine from the University of Virginia. We believe Mr. Link’s extensive medical technology industry and executive experience qualifies him to serve on our board of directors.
Family Relationships
There are no family relationships among any of our directors or executive officers.
Scientific Advisory Board
We have a scientific advisory board, comprised of the following world-renowned scientists with relevant expertise, which helps guide our research and development efforts.
| ● | Claudia Lucchinetti, M.D., Ph.D. | |
| ● | S. Thomas Carmichael, M.D., Ph.D. | |
| ● | Kate Rubins, Ph.D. | |
| ● | Elizabeth Shpall, M.D. | |
| ● | Neil Bhowmick, Ph.D. |
Board of Directors
Our board of directors currently consists of six directors. Our certificate of incorporation provides that, subject to the rights of holders of any series of our preferred stock to elect directors, the number of directors on our board of directors shall be fixed from time to time solely by resolution of the majority of the total number of authorized directors, whether or not there exist any vacancies in previously authorized directorships. Each of our directors serves a term ending on the next annual meeting of our stockholders following such director’s election or appointment, subject to such director’s earlier death, disqualification, resignation or removal.
Pursuant
to our certificate of incorporation, subject to the preferential rights of holders of any series of our preferred stock, any newly created
directorship that results from an increase in the number of directors or any vacancy on our board of directors can only be filled by
the affirmative vote of a majority of the total number of directors then in office, even if less than a quorum, or by a sole remaining
director and cannot be filled by the stockholders. Further, any member of our board of directors or our entire board of directors may
only be removed for cause, and then only by the affirmative vote of the holders of at least 662/3% in voting power of our
stock.
Under our amended and restated certificate of incorporation, which will become effective in connection with the effectiveness of the registration statement of which this prospectus forms a part, upon the effectiveness of the registration statement of which this prospectus forms a part, our board of directors will be divided into three classes, with directors serving staggered three-year terms.
When considering whether directors have the experience, qualifications, attributes or skills, taken as a whole, to enable our board of directors to satisfy its oversight responsibilities effectively in light of our business and structure, the board of directors focuses primarily on each person’s background and experience as reflected in the information discussed in each of the directors’ individual biographies set forth above. We believe that our directors provide an appropriate mix of experience and skills relevant to the size and nature of our business.
| 104 |
Director Independence
Our board of directors has determined that all members of our board of directors, except Pete O’Heeron, are independent directors for purposes of the rules of Nasdaq and the SEC. In making this determination, our board of directors considered the relationships that each non-employee director has with us and all other facts and circumstances that our board of directors deemed relevant, including the beneficial ownership of our common stock by each non-employee director.
Upon the effectiveness of the registration of which this prospectus forms a part, we expect that the composition and functioning of our board of directors and each of our committees will comply with all applicable requirements of Nasdaq and the rules and regulations of the SEC, subject to applicable phase-in periods for committees.
Staggered Board
In accordance with the terms of our amended and restated certificate of incorporation, which will become effective in connection with the effectiveness of the registration statement of which this prospectus forms a part, our board of directors will be divided into three staggered classes of directors and each will be assigned to one of the three classes. At each annual meeting of our stockholders, a class of directors will be elected for a three-year term to succeed the directors of the same class whose terms are then expiring. The terms of the directors will expire upon the election and qualification of successor directors at the annual meeting of shareholders to be held during the years 2024 for Class I directors, 2025 for Class II directors and 2026 for Class III directors.
| ● | Our Class I directors will be Robert Hoffman and Richard Cilento, Jr.; |
| ● | Our Class II directors will be Mathew Link and Victoria Niklas; and |
| ● | Our Class III directors will be Stacy Coen and Pete O’Heeron. |
The division of our board of directors into three classes with staggered three-year terms may delay or prevent stockholder efforts to effect a change of our management or a change in our control.
Board Leadership Structure
Our board of directors is currently chaired by our founder, Pete O’Heeron. Our corporate governance guidelines further provide the flexibility for our board of directors to modify our leadership structure in the future as it deems appropriate.
Committees of our Board of Directors
Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee, each of which operates pursuant to a charter adopted by our board of directors. Our board of directors may also establish other committees from time to time to assist the board of directors. The composition and functioning of all of our committees complies with all applicable requirements of the Sarbanes-Oxley Act, Nasdaq and SEC rules and regulations. Upon our listing on Nasdaq, each committee’s charter will be available on our website at www.fibrobiologics.com.
Audit Committee
The members of our audit committee are Mr. Hoffman, Dr. Niklas, and Mr. Cilento. Mr. Hoffman serves as the chairperson of the committee. Our board of directors has determined that each member of the audit committee is “independent” as that term is defined in Nasdaq rules and has sufficient knowledge in financial and auditing matters to serve on the audit committee. In addition, our board of directors has determined that each member of the audit committee meets the heightened independence requirements for audit committees required under Section 10A of the Exchange Act and related SEC and Nasdaq rules. Our board of directors has determined that Mr. Hoffman is an “audit committee financial expert,” as defined under the applicable rules of the SEC. The audit committee’s responsibilities include:
| ● | appointing, approving the compensation of and assessing the independence of our independent registered public accounting firm; |
| 105 |
| ● | pre-approving auditing and permissible non-audit services, and the terms of such services, to be provided by our independent registered public accounting firm; |
| ● | reviewing the overall audit plan with our independent registered public accounting firm and members of management responsible for preparing our financial statements; |
| ● | reviewing and discussing with management and our independent registered public accounting firm our annual and quarterly financial statements and related disclosures as well as critical accounting policies and practices used by us; |
| ● | coordinating the oversight and reviewing the adequacy of our internal control over financial reporting; |
| ● | establishing policies and procedures for the receipt and retention of accounting-related complaints and concerns; |
| ● | recommending based upon the audit committee’s review and discussions with management and our independent registered public accounting firm whether our audited financial statements shall be included in our annual report on Form 10-K; |
| ● | monitoring the integrity of our financial statements and our compliance with legal and regulatory requirements as they relate to our financial statements and accounting matters; |
| ● | preparing the audit committee report required by SEC rules to be included in our annual proxy statement; |
| ● | reviewing all related person transactions for potential conflict of interest situations and approving all such transactions; and |
| ● | reviewing quarterly earnings releases. |
Compensation Committee
The members of our compensation committee are Mr. Hoffman, Ms. Coen and Mr. Link. Mr. Hoffman serves as the chairperson of the committee. Our board of directors has determined that each member of the compensation committee is “independent” as that term is defined in Nasdaq rules and is a “non-employee director” under Rule 16b-3 under the Exchange Act. In addition, our board of directors has determined that each member of the compensation committee meets the heightened independence requirements for compensation committee purposes under Section 10C of the Exchange Act and related SEC and Nasdaq rules. The compensation committee’s responsibilities include:
| ● | reviewing and approving our philosophy, policies and plans with respect to the compensation of our chief executive officer; |
| ● | making recommendations to our board of directors with respect to the compensation of our chief executive officer and our other executive officers; |
| ● | reviewing and assessing the independence of compensation advisors; |
| ● | overseeing and administering our equity incentive plans; |
| ● | reviewing and making recommendations to our board of directors with respect to director compensation; and |
| ● | preparing the compensation committee reports required by the SEC, including our “compensation discussion and analysis” disclosure. |
| 106 |
Nominating and Corporate Governance Committee
The members of our nominating and corporate governance committee are Ms. Coen, Dr. Niklas and Mr. Link. Ms. Coen serves as the chairperson of the committee. Our board of directors has determined that each member of the nominating and corporate governance committee is “independent” as defined in Nasdaq rules. The nominating and corporate governance committee’s responsibilities include:
| ● | developing and recommending to the board of directors criteria for board and committee membership; |
| ● | establishing procedures for identifying and evaluating board of director candidates, including nominees recommended by shareholders; |
| ● | reviewing the composition of the board of directors to ensure that it is composed of members containing the appropriate skills and expertise to advise us; |
| ● | identifying and screening individuals qualified to become members of the board of directors; |
| ● | recommending to the board of directors the persons to be nominated for election as directors and to each of the board’s committees; |
| ● | developing and recommending to the board of directors a code of business conduct and ethics and a set of corporate governance guidelines; and |
| ● | overseeing the evaluation of our board of directors and management. |
Code of Conduct
We have adopted a written code of business conduct and ethics, that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. In connection with the effectiveness of the registration statement of which this prospectus forms a part, a current copy of the code will be posted on our website at www.fibrobiologics.com. If we make any substantive amendments to, or grant any waivers from, the code of business conduct and ethics for any officer or director, we will disclose the nature of such amendment or waiver on our website or in a current report on Form 8-K.
| 107 |
EXECUTIVE AND DIRECTOR COMPENSATION
Executive Compensation
This section discusses the material components of the executive compensation program for our executive officers who are named in the “—2022 Summary Compensation Table” below. For the fiscal year ended December 31, 2022, our “named executive officers” and their positions were as follows:
| ● | Pete O’Heeron, Chairperson and Chief Executive Officer; | |
| ● | Hamid Khoja, Ph.D., Chief Scientific Officer; and | |
| ● | Mark Andersen, Chief Financial Officer. |
This discussion may contain forward-looking statements that are based on our current plans, considerations, expectations and determinations regarding future compensation programs. Actual compensation programs that we adopt following the completion of this offering may differ materially from the currently planned programs summarized in this discussion. As an “emerging growth company” and a “smaller reporting company,” each as defined under SEC rules, we are not required to include a compensation discussion and analysis section and have elected to comply with the scaled disclosure requirements applicable to emerging growth companies and/or smaller reporting companies.
2022 Summary Compensation Table
The following table represents information regarding the total compensation awarded to, earned by or paid to our named executive officers during the fiscal year ended December 31, 2022:
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Option Awards ($)(2) | All Other Compensation ($)(3) | Total ($) | ||||||||||||||||||
| Pete O’Heeron | 2022 | 600,000 | 300,000 | — | 22,485 | 922,485 | ||||||||||||||||||
| Chairperson and Chief Executive Officer | ||||||||||||||||||||||||
| Hamid Khoja, Ph.D. | 2022 | 300,208 | 148,896 | 23,900 | 20,505 | 493,509 | ||||||||||||||||||
| Chief Scientific Officer | ||||||||||||||||||||||||
| Mark Andersen(1) | 2022 | 189,583 | 81,354 | 20,100 | 88,401 | 379,438 | ||||||||||||||||||
| Chief Financial Officer | ||||||||||||||||||||||||
| (1) | Mark Andersen joined us in June 2022. |
| (2) | In accordance with SEC rules, amounts in this column reflect the aggregate grant date fair value of stock options granted computed in accordance with ASC 718, rather than the amounts paid or realized by the named individual. We provide information regarding the assumptions used to calculate the value of the stock options granted in Note 11 to our audited financial statements included elsewhere in this prospectus. |
| (3) | Amounts in the “All Other Compensation” column consist of the amounts set forth in the table below: |
| Named Executive Officer | 401(k) Plan Matching Contributions ($) |
Healthcare Benefits ($) | Relocation ($) | |||||||||
| Pete O’Heeron | — | 22,485 | — | |||||||||
| Hamid Khoja, Ph.D. | — | 20,505 | — | |||||||||
| Mark Andersen | 6,500 | 20,512 | 61,389 | |||||||||
2022 Salaries
In 2022, our named executive officers received an annual base salary to compensate them for services rendered to us. The base salary payable to each named executive officer is intended to provide a fixed component of compensation reflecting the executive’s skill set, experience, role, and responsibilities.
For fiscal year 2022, Mr. O’Heeron’s annual base salary was $600,000 and Mr. Andersen’s annual base salary was $325,000. Dr. Khoja’s annual base salary was increased from $290,000 to $325,000 during fiscal year 2022.
| 108 |
2022 Bonuses
In fiscal year 2022, Mr. O’Heeron was eligible to receive an annual cash bonus targeted at 50% of his base salary, Dr. Khoja was eligible to earn an annual cash bonus targeted at 35% of base salary, and Mr. Andersen was eligible to earn an annual cash bonus targeted at 35% of base salary, prorated for 2022 based upon the beginning of his employment with us in June 2022.
Each named executive officer was eligible to earn his bonus based on the attainment of pre-established annual company and individual performance objectives, as determined by our board of directors in their discretion. Dr. Khoja was awarded a bonus after his first anniversary in 2022. Future bonuses will be based upon calendar years, with the first calendar year bonus pro rata from the date of his anniversary with us.
Annual bonuses are determined based upon both company performance and individual contributions for the fiscal year and are generally determined and awarded in January of the subsequent year.
Equity Compensation
Dr. Khoja and Mr. Andersen each received commitments in their employment agreements for the equivalent of 7,500 stock options. These options were granted in 2022 after the 2022 Stock Plan (as defined herein) was approved and authorized. Dr. Khoja was also awarded the equivalent of 1,250 shares of non-voting common stock in 2022 prior to establishment of the 2022 Stock Plan. The stock options granted to named executives in 2022 vest 1/3 on the first anniversary date of employment and 1/36th each month thereafter until fully vested, subject to continued service, and will accelerate in full upon the occurrence of a “change in control” of the Company (as defined in the 2022 Stock Plan). For additional information about the 2022 Stock Plan, please see the section titled “—Equity Compensation Plans” below.
Other Elements of Compensation
Retirement Plans
We participate in Insperity’s 401(k) retirement savings plan for our employees, including our named executive officers, who satisfy certain eligibility requirements. Our named executive officers are eligible to participate in the Insperity 401(k) plan on the same terms as other full-time employees. In 2022, contributions made by participants in the 401(k) plan were matched up to a specified percentage of the employee contributions on behalf of the named executive officers. These matching contributions are fully vested as of the date on which the contribution is made. We anticipate that, following the consummation of the Direct Listing, our named executive officers will continue to participate in this Insperity 401(k) plan on the same terms as other full-time employees.
Employee Benefits and Perquisites
Health/Welfare Plans. All of our full-time employees, including our named executive officers, are eligible to participate in Insperity’s health and welfare plans, including:
| ● | medical, dental and vision benefits; | |
| ● | medical and dependent care flexible spending accounts; | |
| ● | short-term and long-term disability insurance; and | |
| ● | life insurance. |
We believe that the employee benefits described above are necessary and appropriate to provide a competitive compensation package to our named executive officers.
| 109 |
Employment Agreements with our Named Executive Officers
Pete O’Heeron Employment Agreement
On December 1, 2023, we entered into an employment agreement with Mr. Peter O’Heeron, pursuant to which Mr. O’Heeron agreed to serve as our President and Chief Executive Officer. Mr. O’Heeron’s employment pursuant to the agreement is “at-will” and is terminable by either party for any reason and with or without notice.
Pursuant to the employment agreement, Mr. O’Heeron is entitled to receive an initial base salary of $600,000, which is to be reviewed annually by the Board of Directors or Compensation Committee but may not be reduced without Mr. O’Heeron’s consent. In addition, the agreement provides that Mr. O’Heeron is eligible to receive an annual performance bonus, as reasonably determined by the Board of Directors or, to the extent delegated by the board, the Compensation Committee, based one or more performance targets annually determined by the board or the committee, provided that to the extent all performance targets are met, the bonus is required to equal not less than 50% of his base salary. The percentage bonus target is to be reviewed periodically by the board or Compensation Committee.
The agreement also provides that Mr. O’Heeron is eligible to participate in the health and welfare benefit plans and programs maintained by us for the benefit of our employees.
Pursuant to the agreement, if Mr. O’Heeron’s employment is terminated by the Company without cause (as defined in the agreement) or by Mr. O’Heeron for good reason (as defined in the agreement), then he will be eligible to receive severance in an amount equal to twelve months’ base salary, paid as if he was still employed during such 12 month period, and the amount of the target bonus that would have been due during such 12 month period (payable 60 days after notice of termination). Additionally, Mr. O’Heeron shall continue to vest options during such 12 month period. If the agreement is terminated for any reason, Mr. O’Heeron is due all compensation earned through the date of termination, including unused and accrued vacation, any unpaid bonus which he is due, and a prorated portion of the bonus which would have accrued for the year of termination (with such bonus amounts being paid at the same time as bonuses are paid to other Company executives).
In the event an involuntary termination of Mr. O’Heeron’s employment occurs during the 12 months following a change in control (as defined in the agreement), or within two months prior to a change in control, or in the event Mr. O’Heeron terminates his employment for any reason not sooner than six months after the occurrence of a change in control, and subject to Mr. O’Heeron entering into a release with the Company, all stock options and stock-based awards held by Mr. O’Heeron, as of the date of notice of such termination are to vest and become exercisable or nonforfeitable.
The agreement contains customary assignment of inventions and confidentiality obligations of Mr. O’Heeron, and a 12 months non-compete/non-solicitation prohibition, following the termination of his employment.
The compensation under the employment agreement (including bonus target) may be increased from time to time, by the Compensation Committee, or the Board of Directors (with the recommendation of the Compensation Committee), which increases do not require the entry into an amended employment agreement.
The Compensation Committee, or the board, with the recommendation of the Compensation Committee, may also pay or grant discretionary cash bonuses or equity bonuses from time to time in their discretion, at any time, in its/their discretion. The equity bonus may be in the form of common stock, stock options or other equity consideration, in such amounts and with such terms as may be determined by the Compensation Committee or the board, with the recommendation of the Compensation Committee, from time to time.
Hamid Khoja, Ph.D. Employment Agreement
We have entered into an employment agreement with Dr. Khoja, dated July 20, 2021, pursuant to which Dr. Khoja serves as our Chief Scientific Officer. Dr. Khoja’s employment pursuant to the agreement is “at-will” and is terminable by either party for any reason and with or without notice.
Pursuant to his agreement, Dr. Khoja is entitled to receive an initial base salary of $290,000, which was increased to $325,000 in 2022. In addition, the agreement provides that Dr. Khoja is eligible to receive an annual performance bonus of up to 35% of his base salary, to be paid based on the achievement of company and individual performance goals. In connection with his entry into the offer letter, Dr. Khoja was granted a stock option award for the equivalent of 7,500 shares of common stock, which vests as to 1/3 of the shares underlying the stock option on the first anniversary of employment date and 1/36th per month thereafter until fully vested, subject to continued employment through the applicable vesting date. Pursuant to the agreement, Dr. Khoja was also paid a one-time cash bonus equal to $15,000 in connection with his commencement of employment and was entitled to payment of up to $45,000 of relocation expenses. The agreement also provides that Dr. Khoja is eligible to participate in the health and welfare benefit plans and programs maintained by us for the benefit of our employees.
Pursuant to the agreement, if Dr. Khoja’s employment is terminated by the Company without cause, then he will be eligible to receive severance in an amount equal to nine months’ base salary.
Mark Andersen Employment Agreement
We have entered into an employment agreement with Mr. Andersen, dated May 20, 2022, pursuant to which Mr. Andersen serves as our Chief Financial Officer. Mr. Andersen’s employment pursuant to the agreement is “at-will” and is terminable by either party for any reason with or without notice.
Pursuant to his agreement, Mr. Andersen is entitled to receive an initial base salary of $325,000. In addition, the agreement provides that Mr. Andersen is eligible to receive an annual performance bonus of up to 35% of his base salary, to be paid based on the achievement of company and individual performance goals. In connection with his entry into the agreement, Mr. Andersen was granted a stock option award for the equivalent of 7,500 shares of common stock, which vests as to 1/3 of the shares underlying the stock option on the first anniversary of employment date and 1/36th per month thereafter, subject to continued employment through the applicable vesting date. Pursuant to the agreement, Mr. Andersen was also paid a one-time cash bonus equal to $15,000 in connection with his commencement of employment and was entitled to payment of up to $45,000 of relocation expenses. The agreement also provides that Mr. Andersen is eligible to participate in the health and welfare benefit plans and programs maintained by us for the benefit of our employees.
Pursuant to the agreement, if Mr. Andersen’s employment is terminated by the Company without cause, then he will be eligible to receive severance in an amount equal to nine months’ base salary.
| 110 |
Equity Compensation Plans
The following summarizes the material terms of the FibroBiologics, Inc. 2022 Stock Plan, or the 2022 Stock Plan.
2022 Stock Plan
Our board of directors adopted on August 10, 2022, and our stockholders approved on August 18, 2022, our 2022 Stock Plan. The 2022 Stock Plan provides for the grant of incentive stock options, nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, and other stock awards. The 2022 Stock Plan, through the grant of stock awards, is intended to help us secure and retain the services of eligible award recipients, provide incentives for such persons to exert maximum efforts for our success and provide a means by which the eligible recipients may benefit from increases in value of our common stock. Through September 30, 2023, we have issued the equivalent of 101,250 options with a strike price of the equivalent of $3.28 per share to employees, directors, and scientific advisory board members, and the equivalent of 3,689,750 options with a strike price of the equivalent of $2.28 per share to employees and directors under the 2022 Stock Plan. In August 2023, a total of 2,500 options with a strike price of $3.28 per share were forfeited. Generally, awards granted by us vest over four years and have an exercise price equal to the estimated fair value of our common stock as determined by our board of directors with consideration given to contemporaneous valuations of our common stock prepared by an independent third-party valuation firm.
As of September 30, 2023, there were the equivalent of 8,711,500 shares available for future issuance under the 2022 Stock Plan.
Outstanding Equity Awards at December 31, 2022
The following table presents information regarding outstanding equity awards held by our named executive officers as of December 31, 2022. Other than the equivalent of 1,250 shares of non-voting common stock awarded to Dr. Khoja in 2022 prior to establishment of the 2022 Stock Plan, all awards were granted under our 2022 Stock Plan.
| Name |
Number of securities underlying unexercised options (#) exercisable |
Number of securities underlying unexercised options (#) unexercisable |
Option exercise price ($) |
Option expiration date |
||||||||||||
| Pete O’Heeron | — | — | — | — | ||||||||||||
| Hamid Khoja, Ph.D. | 3,542 | 3,958 | 3.28 | September 25, 2032 | ||||||||||||
| Mark Andersen | — | 7,500 | 3.28 | September 25, 2032 | ||||||||||||
Director Compensation
Non-employee Director Compensation Table
The following table presents the total compensation for each person who served as a non-employee member of our board of directors during the fiscal year ended December 31, 2022. Other than as set forth in the table and described more fully below, we did not pay any compensation, make any equity awards or non-equity awards to, or pay any other compensation to any of the non-employee members of our board of directors in 2022 for their services as members of our board of directors. Pete O’Heeron, our Chairperson and Chief Executive Officer, received no additional compensation for his service as a director. See the section titled “Executive Compensation” for more information on the compensation paid to or earned by O’Heeron as an employee for the year ended December 31, 2022.
| Name | Fees Earned or Paid in Cash ($) | Stock Awards ($) (1)(3) | Option Awards ($)(2)(3) | Total ($) | ||||||||||||
| Robert Hoffman | 50,000 | 24,600 | 12,800 | 87,400 | ||||||||||||
| Victoria Niklas | 43,000 | 24,600 | 12,800 | 80,400 | ||||||||||||
| Richard Cilento, Jr. | 43,000 | 24,600 | 12,800 | 80,400 | ||||||||||||
| Stacy Coen | 41,000 | 24,600 | 12,800 | 78,400 | ||||||||||||
| Matthew Link | 41,000 | 24,600 | 12,800 | 78,400 | ||||||||||||
| (1) | In January 2022, each of our non-employee directors was awarded the equivalent of 7,500 shares of stock. | |
| (2) | In September 2022, each of our non-employee directors was granted the equivalent of 5,000 stock options with an exercise price of the equivalent of $3.28 per share. | |
| (3) | The amounts reported represent the aggregate grant date fair value of the stock and stock options awarded to the non-employee directors during fiscal year 2022, calculated in accordance with ASC Topic 718. Such grant date fair value does not take into account any estimated forfeitures. The assumptions used in calculating the grant date fair value of the awards reported in this column are set forth in the notes to our financial statements included elsewhere in this prospectus. The amounts reported in this column reflect the accounting cost for the stock and stock options and do not correspond to the actual economic value that may be received upon exercise of the stock options or any sale of any of the underlying shares of common stock. |
As of December 31, 2022, the non-employee members of our board of directors held the following aggregate number of unexercised options:
| Name | Number of Securities Underlying Unexercised Options |
|||
| Robert Hoffman | 5,000 | |||
| Victoria Niklas | 5,000 | |||
| Richard Cilento | 5,000 | |||
| Stacy Coen | 5,000 | |||
| Matthew Link | 5,000 | |||
Except as set forth above, no non-employee member of our board of directors held unexercised options or unvested shares of our common stock as of December 31, 2022.
| 111 |
Non-Employee Director Compensation Policy
Our board of directors has adopted a non-employee director compensation policy that will continue upon the effectiveness of the registration statement of which this prospectus is a part. The policy is designed to enable us to attract and retain, on a long-term basis, highly qualified non-employee directors. Under the policy, each director who is not an employee will be paid cash compensation from and after the completion of this offering as set forth below:
| Position | Annual Retainer | |||
| Board of Directors: | ||||
| Members (other than chair) | $ | 35,000 | ||
| Audit Committee: | ||||
| Members (other than chair) | $ | 8,000 | ||
| Retainer for chair | $ | 10,000 | ||
| Compensation Committee: | ||||
| Members (other than chair) | $ | 6,000 | ||
| Retainer for chair | $ | 10,000 | ||
| Nominating and Corporate Governance Committee: | ||||
| Members (other than chair) | $ | 5,000 | ||
| Retainer for chair | $ | 10,000 | ||
In addition, the non-employee director compensation policy provides that, upon initial election to our board of directors, each non-employee director will be granted an equity award the equivalent of 7,500 shares of common stock, or the Initial Grant. Furthermore, on the date of each of our annual meeting of stockholders, each non-employee director who continues as a non-employee director following such meeting will be granted an annual equity award of stock options, to purchase the equivalent of 5,000 shares, or the Annual Grant. The Annual Grant will vest in full upon the earlier of (i) the first anniversary of the date of grant or (ii) the date of the next annual meeting; provided, however, that all vesting shall cease if the director resigns from the board of directors or otherwise ceases to serve as a director, unless the board of directors determines that the circumstances warrant continuation of vesting. In addition, all vested options remain exercisable for 12 months if the director resigns from the board of directors or otherwise ceases to serve as a director. Notwithstanding the foregoing, if an outside director was initially elected to the board of directors within 12 months preceding the annual meeting, then such outside director shall receive an Annual Grant that is pro-rated on a monthly basis for time serving as an outside director.
| 112 |
CERTAIN RELATIONSHIPS AND RELATED PERSON TRANSACTIONS
The following is a summary of transactions or series of transactions since inception, or currently proposed transactions or series of transactions, to which we were, or will be, a party, in which the amount involved exceeded, or will exceed, $120,000, and in which any of our directors, executive officers, or to our knowledge, beneficial owners of 5% or more of our capital stock, or 5%+ Security Holders, or any member of the immediate family of, or entities affiliated with, any of the foregoing persons, had, or will have, a direct or indirect material interest.
Series A Preferred Stock
In May 2021, as part our formation, we issued the equivalent of 8,750,000 shares of our Series A Preferred Stock to FibroGenesis in exchange for a Patent Assignment Agreement, which assigns certain patents/applications to us, and an Intellectual Property Cross-License Agreement, which provides to us an exclusive license within defined fields of use for patents/applications retained by FibroGenesis and provides to FibroGenesis an exclusive license to the patents/applications assigned to FibroBiologics for all other fields of use.
In connection with the Direct Listing, all of our then outstanding Series A Preferred Stock will be automatically canceled, without the payment of additional consideration by or to the holder thereof.
FibroGenesis Loans
In July 2022, we loaned $300,000 to FibroGenesis at 0% interest and one year maturity date. In October 2022, we loaned an additional $60,000 to FibroGenesis at 0% interest and one year maturity. The $60,000 was fully repaid in December 2022 and the $300,000 was fully repaid in April 2023.
ROFN Agreement
In January 2023, we entered into an Agreement Regarding Right of First Negotiation with FibroGenesis, or the ROFN Agreement. In exchange for FibroGenesis’ consent to amend our certificate of incorporation to (i) eliminate upon our underwritten initial public offering or the direct listing of our common stock on a securities exchange (which we collectively refer to as an IPO) or sale of our company, the liquidation preference for the Series A Preferred Stock, (ii) make the Series B Preferred Stock liquidation preference equal to Series A Preferred Stock and (iii) to provide that upon an IPO or sale of our company, the Series A Preferred Stock will be canceled for no consideration, we agreed to pay to FibroGenesis 15% of the gross proceeds from any equity investments in us prior to an IPO or sale of our company. In addition, we received a five-year right of first negotiation if FibroGenesis decides to license externally any of its technology. Through September 30, 2023, we have paid a total of $2.6 million to FibroGenesis under the ROFN Agreement based upon gross proceeds from equity investments received through September 30, 2023.
2021 and 2022 Convertible Notes
In December 2021, we issued and sold to investors, some of whom hold more than 5% shares, in a private placement $1.3 million of our convertible promissory notes, or the 2021 Notes. The 2021 Notes bore interest at an initial interest rate of 6.0% per annum and would have automatically converted into shares of our common stock in the event of a qualified financing. The conversion price of the 2021 Notes was equal to $200.0 million divided by the total number of equity interests prior to the dilution from the offering. The 2021 Notes were unsecured and subordinated in right of payment to the prior payment in full to all of our commercial finance lenders, insurance companies, lease financing institutions or other lending institutions approved by our board of directors and regularly engaged in the business of lending money. In April 2023, $1.3 million of these notes were converted into shares of our Series B Preferred Stock and none of the 2021 Notes are outstanding.
In January 2022 and April 2022, we issued and sold to investors, some of whom hold more than 5% of shares, in a private placement $0.35 million and $3.95 million, respectively, of our convertible promissory notes, or the 2022 Notes. The 2022 Notes bore interest at an initial interest rate of 6.0% per annum, had a one-year maturity, and could have been converted at the holder’s request into shares of our common stock in the event of a qualified financing. The conversion price of the 2022 Notes was the lesser of (i) a 15% discount to the offering price of our common stock in the event of an IPO or (i) the quotient of $200.0 million divided by total equity interests prior to the dilution from the offering. The 2022 Notes were unsecured and subordinated in right of payment to the prior payment in full to all of our commercial finance lenders, insurance companies, lease financing institutions or other lending institutions approved by our board of directors and regularly engaged in the business of lending money. In February 2023 through June 2023, $4.3 million of these notes were converted into shares of our Series B Preferred Stock and none of the 2022 Notes are outstanding.
Equity and Compensation Arrangements
We adopted on August 10, 2022, and our stockholders approved on August 18, 2022, our 2022 Stock Plan, or the 2022 Plan. The 2022 Plan provides for the grant of incentive stock options, non-statutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, and other stock awards. We issued in 2022 a total of the equivalent of 101,250 options with an exercise price the equivalent of $3.28 per share to employees, directors, and scientific advisory board members under the 2022 Plan. In February 2023, we issued an additional equivalent of 3,689,750 options with an exercise price the equivalent of $2.28 per share to employees and directors. Generally, awards granted by the Company vest over three years and have an exercise price equal to the estimated fair value of the common stock as determined by our board of directors with consideration given to contemporaneous valuations of our common stock prepared by an independent third-party valuation firm.
| 113 |
PRINCIPAL AND REGISTERED STOCKHOLDERS
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth as of , 2023:
| ● | certain information regarding the beneficial ownership of our voting securities (being our voting common stock and our Series C Preferred Stock) as of , 2023 by (i) each person or group of affiliated persons known by us to be the beneficial owner of more than 5% of our voting securities, (ii) each of our executive officers, (iii) each of our directors and (iv) all of our directors and executive officers as a group. Except as otherwise indicated, all persons listed below have (i) sole voting power and investment power with respect to their common stock, except to the extent that authority is shared by spouses under applicable law, and (ii) record and beneficial ownership with respect to their common stock; and |
| ● | the number of shares of our common stock held by and registered for resale by means of this prospectus for the Registered Stockholders. |
The Registered Stockholders include (i) our affiliates and certain other stockholders with “restricted securities” (as defined in Rule 144 under the Securities Act) who, because of their status as affiliates pursuant to Rule 144 or because they acquired their common stock from an affiliate or us within the prior 12 months, would be unable to sell their securities pursuant to Rule 144 until we have been subject to the reporting requirements of Section 13 or Section 15(d) of the Exchange Act for a period of at least 90 days and (ii) our employees. The Registered Stockholders may, or may not, elect to sell their common stock covered by this prospectus, as and to the extent they may determine. The Registered Stockholders may offer, sell or distribute all or a portion of the shares of common stock hereby registered publicly or through private transactions at prevailing market prices or at negotiated prices. The Registered Stockholders may elect to sell their shares in connection with this Direct Listing and in market transactions following this Direct Listing. As such, we will have no input if and when any Registered Stockholder may, or may not, elect to sell their common stock or the prices at which any such sales may occur. See “Plan of Distribution.”
Information concerning the Registered Stockholders may change from time to time and any changed information will be set forth in supplements to this prospectus, if and when necessary. Because the Registered Stockholders may sell all, some, or none of the common stock covered by this prospectus, we cannot determine the number of common stock that will be sold by the Registered Stockholders, or the amount or percentage of shares of common stock that will be held by the Registered Stockholders upon consummation of any particular sale. In addition, the Registered Stockholders listed in the table below may have sold, transferred, or otherwise disposed of, or may sell, transfer, or otherwise dispose of, at any time and from time to time, our common stock in transactions exempt from the registration requirements of the Securities Act, after the date on which they provided the information set forth in the table below.
The Registered Stockholders are not entitled to any registration rights with respect to the common stock. However, we currently intend to use our reasonable efforts to keep the registration statement effective for a period of 90 days after the effectiveness of the registration statement. We are not party to any arrangement with any Registered Stockholder or any broker-dealer with respect to sales of common stock by the Registered Stockholders. However, we will engage a financial advisor with respect to certain other matters relating to our listing. See “Plan of Distribution.”
| 114 |
In accordance with the rules of the SEC, beneficial ownership includes voting or investment power with respect to securities and includes the common stock issuable pursuant to options and warrants that are exercisable or settled within 60 days of , 2023. Shares of common stock issuable pursuant to options and warrants are deemed outstanding for computing the percentage of the class beneficially owned by the person holding such securities but are not deemed outstanding for computing the percentage of the class beneficially owned by any other person. The percentage of beneficial ownership for the following table is based on total shares of common stock outstanding as of , 2023.
In the table below, the percentage of beneficial ownership prior to the effectiveness of the registration statement of which this prospectus forms a part is based on, as applicable: (i) 32,492,068 shares of our common stock outstanding as of , 2023, after giving effect to the Reverse Stock Split and (a) the automatic cancellation, in connection with the Direct Listing, of all of our outstanding Series A Preferred Stock, (b) the automatic conversion, in connection with the Direct Listing, of all of our outstanding non-voting common stock, on a one-for-one basis, into an aggregate of 28,230,842 shares of our common stock, (c) the automatic conversion, in connection with the Direct Listing, of all of our outstanding Series B Preferred Stock, on a one-for-one basis, into an aggregate of 4,171,445 shares of our common stock and (d) the automatic conversion, in connection with the Direct Listing, of all our outstanding Series B-1 Preferred Stock into an aggregate of 89,781 shares of our common stock; and (ii) 2,500 shares of our Series C Preferred Stock outstanding as of , 2023, after giving effect to the Reverse Stock Split.
Each share of our Series C Preferred Stock will be entitled to 13,000 votes per share upon the Direct Listing. The percentage of total voting power in the table below is based on, after giving effect to the transactions described in clause (i) and (ii) in the immediately preceding paragraph and the 13,000 votes per share of Series C Preferred Stock upon the Direct Listing, the sum of (i) 32,492,068 votes, being the total number of votes associated with the 32,492,068 shares of our common stock (with each share of common stock having one vote) and (ii) 32,500,000 votes, being the total number of votes associated with the 2,500 shares of Series C Preferred Stock (with each share of Series C Preferred Stock having 13,000 votes).
FibroGenesis is the beneficial owner of all of our outstanding Series A Preferred Stock (being an aggregate of 8,750,000 shares in number, after giving effect to the Reverse Stock Split), all of which outstanding Series A Preferred Stock will be automatically cancelled, without consideration, in connection with the Direct Listing. Other than such Series A Preferred Stock, all of which will be cancelled as aforesaid, FibroGenesis does not own any other shares of our capital stock.
The Registered Stockholders have not, nor have they within the past three years had, any position, office, or other material relationship with us, other than as disclosed in this prospectus. See “Management’s Discussion & Analysis of Financial Results and Condition” and “Certain Relationships and Related Party Transactions” for further information regarding the Registered Stockholders. Unless otherwise indicated, the business address of each of the individuals and entities named below is c/o FibroBiologics, Inc., 455 E. Medical Center, Blvd., Suite 300, Houston, Texas 77598.
| Beneficial Ownership Prior to the Effectiveness of the Registration Statement | ||||||||||||||||||||||||
| Common Stock | Series C Preferred Stock | Percentage of Total | Shares of Common Stock Being Registered Pursuant to | |||||||||||||||||||||
| Name and address of Beneficial Owner | Shares | % | Shares | % | Voting Power(1) | this Prospectus | ||||||||||||||||||
| 5% Stockholders: | ||||||||||||||||||||||||
| Golden Knight Incorporated, L.P.(2) | 2,125,001 | 6.5 | % | — | — | 3.3 | % | 218,351 | ||||||||||||||||
| Dan and Pam Linscomb(3) | 1,627,219 | 5.0 | % | — | — | 2.5 | % | 1,627,219 | ||||||||||||||||
| Executive Officers and Directors | ||||||||||||||||||||||||
| Pete O’Heeron, MSHA(4) | 6,048,147 | 18.6 | % | 2,500 | 100 | % | 59.3 | % | — | |||||||||||||||
| Mark Andersen, CPA CFA | — | — | — | — | — | — | ||||||||||||||||||
| Hamid Khoja, Ph.D.(5) | 1,250 | * | — | — | * | — | ||||||||||||||||||
| Robert Hoffman, CPA (inactive)(6) | 7,500 | * | — | — | * | — | ||||||||||||||||||
| Victoria Niklas, M.D.(7) | 7,500 | * | — | — | * | — | ||||||||||||||||||
| Richard Cilento, Jr., MBA(8) | 93,225 | * | — | — | * | — | ||||||||||||||||||
| Stacy Coen, MBA(9) | 7,500 | * | — | — | * | — | ||||||||||||||||||
| Matthew Link(10) | 7,500 | * | — | — | * | — | ||||||||||||||||||
| Directors and Executive Officers as a Group (8 persons)(11) | 6,172,622 | 19.0 | % | 2,500 | 100 | % | 59.5 | % | — | |||||||||||||||
| Other Registered Stockholders: | ||||||||||||||||||||||||
| Non-Executive Officer Employees, Consultants and Service Providers | — | — | — | — | — | — | ||||||||||||||||||
| Keith Denner Rev Trust(12) | 677,367 | 2.1 | % | — | — | 1.0 | % | 507,245 | ||||||||||||||||
| Benny Brown(13) | 565,155 | 1.7 | % | — | — | * | 455,856 | |||||||||||||||||
| Collins Partners Ltd(14) | 509,489 | 1.6 | % | — | — | * | 40,065 | |||||||||||||||||
| Michael M. Lavor(15) | 497,754 | 1.5 | % | — | — | * | 5,873 | |||||||||||||||||
| Global Energy Source Inc.(16) | 465,744 | 1.4 | % | — | — | * | 272,500 | |||||||||||||||||
| Mod Capital LLC(17) | 403,799 | 1.2 | % | — | — | * | 4,438 | |||||||||||||||||
| Mustasim Rumi(18) | 350,280 | 1.1 | % | — | — | * | 38,462 | |||||||||||||||||
| Mark Monical(19) | 313,223 | 1.0 | % | — | — | * | 183,956 | |||||||||||||||||
| Philip P. Lovell and Elizabeth G. Lovell(20) | 212,803 | * | — | — | * | 154,890 | ||||||||||||||||||
| James P. Doherty, III Trust Dated Nov 14, 2000(21) | 189,868 | * | — | — | * | 91,250 | ||||||||||||||||||
| Barchil Ltd Partnership(22) | 66,077 | * | — | — | * | 66,077 | ||||||||||||||||||
| David M Kavanagh(23) | 251,574 | * | — | — | * | 92,557 | ||||||||||||||||||
| Megan Doherty(24) | 45,000 | * | — | — | * | 45,000 | ||||||||||||||||||
| Sean Ryan Trust dtd 3/15/07(25) | 106,579 | * | — | — | * | 44,822 | ||||||||||||||||||
| Slade Lewis(26) | 99,359 | * | — | — | * | 44,436 | ||||||||||||||||||
| Steven Caperton(27) | 98,600 | * | — | — | * | 62,167 | ||||||||||||||||||
| Anthony J. Lydon(28) | 105,044 | * | — | — | * | 44,379 | ||||||||||||||||||
| Darrell Jones(29) | 135,749 | * | — | — | * | 38,462 | ||||||||||||||||||
| Lynn Davis DDS(30) | 62,735 | * | — | — | * | 33,332 | ||||||||||||||||||
| Avatar Investments, LLC(31) | 132,164 | * | — | — | * | 24,424 | ||||||||||||||||||
| Shanala Jap Investment Services LLLP(32) | 25,934 | * | — | — | * | 25,934 | ||||||||||||||||||
| Richard Keathley(33) | 29,576 | * | — | — | * | 22,225 | ||||||||||||||||||
| Sandra Lewis(34) | 22,218 | * | — | — | * | 22,218 | ||||||||||||||||||
| John Hillis(35) | 79,081 | * | — | — | * | 22,190 | ||||||||||||||||||
| StartEngine Capital LLC(36) | 22,148 | * | — | — | * | 22,148 | ||||||||||||||||||
| Jay Dempsey(37) | 21,779 | * | — | — | * | 21,779 | ||||||||||||||||||
| George Nesemeier(38) | 75,486 | * | — | — | * | 19,231 | ||||||||||||||||||
| J. David Taormino(39) | 44,686 | * | — | — | * | 19,231 | ||||||||||||||||||
| Ryan Tabloff(40) | 18,677 | * | — | — | * | 18,677 | ||||||||||||||||||
| Michael Jones(41) | 46,618 | * | — | — | * | 17,752 | ||||||||||||||||||
| William Austin(42) | 17,752 | * | — | — | * | 17,752 | ||||||||||||||||||
| Michael Moses(43) | 17,752 | * | — | — | * | 17,752 | ||||||||||||||||||
| Scott Dluzen(44) | 16,346 | * | — | — | * | 16,346 | ||||||||||||||||||
| Lynda Ludwig(45) | 19,949 | * | — | — | * | 11,199 | ||||||||||||||||||
| GNGI Investment Services LLLP(46) | 12,774 | * | — | — | * | 12,774 | ||||||||||||||||||
| Victor A Estrada(47) | 143,543 | * | — | — | * | 9,764 | ||||||||||||||||||
| Jerry Collins(48) | 8,876 | * | — | — | * | 8,876 | ||||||||||||||||||
| Ivan I Smith III(49) | 25,412 | * | — | — | * | 8,506 | ||||||||||||||||||
| James Sage(50) | 8,506 | * | — | — | * | 8,506 | ||||||||||||||||||
| Chris McCall(51) | 8,387 | * | — | — | * | 8,387 | ||||||||||||||||||
| Gladys E. Pierson(52) | 10,644 | * | — | — | * | 10,644 |
||||||||||||||||||
| Schnoy, LLC(53) | 38,825 | * | — | — | * | 8,188 | ||||||||||||||||||
| Marshall A. Lewis(54) | 8,136 | * | — | — | * | 8,136 | ||||||||||||||||||
| John Mai(55) | 7,692 | * | — | — | * | 7,692 | ||||||||||||||||||
| Barbara Peraskevas(56) | 7,316 | * | — | — | * | 7,316 | ||||||||||||||||||
| Sabio Viloria(57) | 12,982 | * | — | — | * | 7,101 | ||||||||||||||||||
| Chad Burns(58) | 7,101 | * | — | — | * | 7,101 | ||||||||||||||||||
| Greg Maher(59) | 21,174 | * | — | — | * | 6,472 | ||||||||||||||||||
| Robert J Anthony(60) | 7,096 | * | — | — | * | 7,096 | ||||||||||||||||||
| Steve Rice(61) | 5,104 | * | — | — | * | 5,104 | ||||||||||||||||||
| Rich Sangberg(62) | 4,863 | * | — | — | * | 4,863 | ||||||||||||||||||
| Charles Cadenhead(63) | 4,808 | * | — | — | * | 4,808 | ||||||||||||||||||
| RoboSpine LLC(64) | 4,623 | * | — | — | * | 4,623 | ||||||||||||||||||
| Ted Fowler(65) | 52,421 | * | — | — | * | 4,582 | ||||||||||||||||||
| J. Dickson Mappus(66) | 29,893 | * | — | — | * | 4,438 | ||||||||||||||||||
| Mark Drinkwater(67) | 17,166 | * | — | — | * | 4,438 | ||||||||||||||||||
| Darrin Shandley(68) | 4,438 | * | — | — | * | 4,438 | ||||||||||||||||||
| Mary Taormino(69) | 4,438 | * | — | — | * | 4,438 | ||||||||||||||||||
| Wichuta Prochnow(70) | 4,068 | * | — | — | * | 4,068 | ||||||||||||||||||
| Bart Pasternak(71) | 3,938 | * | — | — | * | 3,938 | ||||||||||||||||||
| Charles Read(72) | 3,883 | * | — | — | * | 3,883 | ||||||||||||||||||
| Sheryl Moses(73) | 3,872 | * | — | — | * | 3,872 | ||||||||||||||||||
| Robert Myers Retirement Plan-Roth(74) | 3,750 | * | — | — | * | 3,750 | ||||||||||||||||||
| Willam Ford(75) | 41,865 | * | — | — | * | 3,551 | ||||||||||||||||||
| Gloria Lopez(76) | 1,480 | * | — | — | * | 1,480 | ||||||||||||||||||
| Donna L Brown(77) | 740 | * | — | — | * | 740 | ||||||||||||||||||
| Jason D Sellers(78) | 5,925 | * | — | — | * | 5,925 | ||||||||||||||||||
| All Other Registered Stockholders(79) | 612,747 | 1.9 | % | — | — | % | * | % | 236,533 | |||||||||||||||
| Total Number of Shares Being Registered | 4,806,226 | |||||||||||||||||||||||
* Less than 1%.
| (1) | After giving effect to the rights of the Series C Preferred Stock, upon the Direct Listing, to 13,000 votes per share. |
| (2) | The number of shares of common stock held consists of an aggregate of (i) 1,906,650 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 1,906,650 shares of common stock and (ii) 218,351 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 218,351 shares of common stock. Michael F. Newlin and Cindy L. Newlin, as General Partners of Golden Knight Incorporated, L.P., share discretionary authority to vote and dispose of the shares directly held by Golden Knight Incorporated, L.P. and may be deemed to be the beneficial owners of such shares. The address for Golden Knight Incorporated, L.P. is 3773 Howard Hughes Pkwy, Suite 500S, Las Vegas, NV 89189-6014. |
| (3) | The number of shares of common stock held consists of 1,627,219 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 1,627,219 shares of common stock. The address for Dan and Pam Linscomb is 5110 San Felipe St, #374, Houston, TX 77056. |
| (4) | The number of shares of common stock held consists of 6,048,147 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 6,048,147 shares of common stock. The 2,500 shares of Series C Preferred Stock held constitute the maximum number of Series C Preferred Stock we are authorized to issue. Upon the Direct Listing, each share of Series C Preferred Stock will be entitled to 13,000 votes. For as long as they remain outstanding, the Series C Preferred Stock will be subject to an irrevocable proxy issued by Pete O’Heeron in favor and for the benefit of our board of directors, as more particularly described in this prospectus. |
| (5) | The number of shares of common stock held consists of 1,250 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 1,250 shares of common stock. |
| (6) | The number of shares of common stock held consists of 7,500 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 7,500 shares of common stock. |
| (7) | The number of shares of common stock held consists of 7,500 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 7,500 shares of common stock. |
| (8) | The number of shares of common stock held consists of 93,225 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 93,225 shares of common stock. |
| (9) | The number of shares of common stock held consists of 7,500 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 7,500 shares of common stock |
| (10) | The number of shares of common stock held consists of 7,500 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 7,500 shares of common stock. |
| (11) | The number of shares of common stock held consists of 6,172,622 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 6,172,622 shares of common stock. The 2,500 shares of Series C Preferred Stock held constitute the maximum number of Series C Preferred Stock we are authorized to issue. Upon the Direct Listing, each share of Series C Preferred Stock will be entitled to 13,000 votes. For as long as they remain outstanding, the Series C Preferred Stock will be subject to an irrevocable proxy issued by Pete O’Heeron in favor and for the benefit of our board of directors, as more particularly described in this prospectus. |
| (12) | The number of shares of common stock held consists of (i) 170,122 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into an aggregate of 170,122 shares of common stock and (ii) 507,245 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 507,245 shares of common stock. The address for the Keith Denner Rev Trust is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (13) | The number of shares of common stock held consists of an aggregate of (i) 109,299 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 109,299 shares of common stock and (ii) 455,856 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 455,856 shares of common stock. The address for Benny Brown is 4338 N. Chapel Rd., Franklin, TN 37067. |
| (14) | The number of shares of common stock held consists of an aggregate of (i) 469,424 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 469,424 shares of common stock, (ii) 20,711 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 20,711 shares of common stock, and (iii) 19,354 shares of Series B-1 Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 19,354 shares of common stock. The address for Collins Partners Ltd is 5000 Burnet Rd, Austin, TX 78756. |
| (15) | The number of shares of common stock held in aggregate by Michael A. Lavor, Wildcat Navy LLLP, and Mission Management & Trust Co. FBO Michael M. Lavor SEP IRA with Michael A. Lavor as beneficiary consists of an aggregate of (i) 491,881 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 491,881 shares of common stock and (ii) 5,873 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 5,873 shares of common stock. The address for Michael A. Lavor is 3650 N Camino Ojo DeAgua, Tucson, AZ 85749. |
| (16) | The number of shares of common stock held consists of 465,744 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 465,744 shares of common stock. The address for Global Energy Source Inc is 6233 N. 75th St, Scottsdale, AZ 85250. |
| (17) | The number of shares of common stock held consists of an aggregate of (i) 399,361 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 399,361 shares of common stock and (ii) 4,438 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,438 shares of common stock. The address for Mod Capital LLC is 14840 Jolenta Ln, Elm Grove, WI 53122. |
| (18) | The number of shares of common stock held in aggregate by Mustasim Rumi, MDR Strategic Investments, LLC, Quest IRA FBO Mustasim Rumi, Sarah V Investments, LLC, and Vendal-Rumi Irrevocable Trust with Mustasim Rumi as beneficiary consists of an aggregate of (i) 311,818 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 311,818 shares of common stock and (ii) 38,462 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 38,462 shares of common stock. The address for Mustasim Rumi is 5919 Bold Ruler Way, Austin, TX 78746. |
| (19) | The number of shares of common stock held consists of an aggregate of (i) 129,267 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 129,267 shares of common stock and (ii) 183,956 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 183,956 shares of common stock. The address for Mark Monical is 78 Rosewood St, Lake Jackson, TX 77566. |
| (20) | The number of shares of common stock held in aggregate by The Lovell Family Trust dated 3/12/02 and Vantage Roth IRA FBO Philip Lovell with Philip P. Lovell and Elizabeth G. Lovell as beneficiaries consists of an aggregate of (i) 194,163 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 194,163 shares of common stock and (ii) 18,640 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 18,640 shares of common stock. The address for Philip P. Lovell and Elizabeth G. Lovell is 4601 E. Foothill Drive, Paradise Valley, AZ 85253. |
| (21) | The number of shares of common stock held consists of 189,868 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 189,868 shares of common stock. The address for James P. Doherty, III Trust Dated Nov 14, 2000 is 107 Whisper Dunes Dr., Michigan City, IN 46360. |
| (22) | The number of shares of common stock held consists of 66,077 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 66,077 shares of common stock. The address for Barchil Ltd Partnership is PO Box 11739, Fort Worth, TX 76110. |
| (23) | The number of shares of common stock held in aggregate by David Kavanagh, David M Kavanagh Trust, Dearborn Capital Management, Ltd., and David Kavanagh 2010 Trust with David M Kavanagh and family as beneficiary consists of an aggregate of (i) 159,017 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 159,017 shares of common stock and (ii) 92,557 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 92,557 shares of common stock. The address for David M Kavanagh is 2001 Knollwood Rd, Lake Forest, IL 60045. |
| (24) | The number of shares of common stock held consists of 45,000 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 45,000 shares of common stock. The address for Megan Doherty is 145 Montgomery St, Glencoe IL 60022. |
| (25) | The number of shares of common stock held consists of an aggregate of (i) 61,757 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 61,757 shares of common stock and (ii) 44,822 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 44,822 shares of common stock. The address for Sean Ryan Trust dtd 3/15/07 is 1425 E Pinto Ct, Gilbert, AZ 85233. |
| (26) | The number of shares of common stock held consists of an aggregate of (i) 54,923 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 54,923 shares of common stock and (ii) 44,436 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 44,436 shares of common stock. The address for Slade Lewis is 501 Hunters Lane, Friendswood, TX 77546. |
| (27) | The number of shares of common stock held in aggregate by Steven Caperton and Painted River Royalties LLC with Steven Caperton as beneficiary consists of an aggregate of (i) 36,433 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 36,433 shares of common stock and (ii) 62,167 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 62,167 shares of common stock. The address for Steven Caperton is 1855 Painted River Rd, Valley Mills, TX 76689. |
| (28) | The number of shares of common stock held in aggregate by Anthony J. Lydon and Anthony J. Lydon Living Trust with Anthony J. Lydon as beneficiary consists of an aggregate of (i) 60,665 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 60,665 shares of common stock and (ii) 44,379 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 44,379 shares of common stock. The address for Anthony J. Lydon is 4300 E. Camelback Rd, Ste 100, Phoenix, AZ 85018. |
| (29) | The number of shares of common stock held in aggregate by Darrell Jones and Darrell Matthew Jones Trust with Darrell Jones as beneficiary consists of an aggregate of (i) 97,287 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 97,287 shares of common stock and (ii) 38,462 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 38,462 shares of common stock. The address for Darrell Jones is PO Box 68, Sedan, Kansas 67361. |
| (30) | The number of shares of common stock held consists of an aggregate of (i) 29,403 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 29,403 shares of common stock and (ii) 33,332 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 33,332 shares of common stock. The address for Lynn Davis DDS is 19 Ellis Rd, League City, TX 77573. |
| (31) | The number of shares of common stock held in aggregate by Avatar Investments, LLC, with Galen Miler as beneficiary consists of an aggregate of (i) 107,740 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 107,740 shares of common stock and (ii) 24,424 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 24,424 shares of common stock. The address for Galen Miler is 20539 N. Bear Canyon Ct., Surprise, AZ 85387. |
| (32) | The number of shares of common stock held consists of 25,934 shares of Series B-1 Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 25,934 shares of common stock. The address for Shanala Jap Investment Services LLLP is 5995 E Grant Rd, Suite 200, Tucson, AZ 85712. |
| (33) | The number of shares of common stock held consists of an aggregate of (i) 7,351shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 7,351 shares of common stock and (ii) 22,225 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 22,225 shares of common stock. The address for Richard Keathley is 104 Anderson Ranch Ln, Friendswood, TX 77546. |
| (34) | The number of shares of common stock held consists of 22,218 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 22,218 shares of common stock. The address for Sandra Lewis is 204 Hidden Pines Ct, League City, TX 77573. |
| (35) | The number of shares of common stock held consists of an aggregate of (i) 56,891 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 56,891 shares of common stock and (ii) 22,190 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 22,190 shares of common stock. The address for John Hillis is 400 Russell Park, Davis, CA 95616. |
| (36) | The number of shares of common stock held consists of 22,148 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 22,148 shares of common stock. The address for StartEngine Capital LLC is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (37) | The number of shares of common stock held consists of 21,779 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 21,779 shares of common stock. The address for Jay Dempsey is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (38) | The number of shares of common stock held consists of an aggregate of (i) 56,255 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 56,255 shares of common stock and (ii) 19,231 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 19,231 shares of common stock. The address for George Nesemeier is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (39) | The number of shares of common stock held consists of an aggregate of (i) 25,455 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 25,455 shares of common stock and (ii) 19,231 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 19,231 shares of common stock. The address for J. David Taormino is 429 F Street, #5, Davis, CA 95616. |
| (40) | The number of shares of common stock held consists of 18,677 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 18,677 shares of common stock. The address for Ryan Tabloff is 7343 E Claremont, Scottsdale, AZ 85250. |
| (41) | The number of shares of common stock held consists of an aggregate of (i) 28,866 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 28,866 shares of common stock and (ii) 17,752 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 17,752 shares of common stock. The address for Michael Jones is 234 Road 20, Sedan, KS 67361. |
| (42) | The number of shares of common stock held consists of 17,752 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 17,752 shares of common stock. The address for William Austin is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (43) | The number of shares of common stock held consists of 17,752 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 17,752 shares of common stock. The address for Michael Moses is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (44) | The number of shares of common stock held consists of 16,346 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 16,346 shares of common stock. The address for Scott Dluzen is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (45) | The number of shares of common stock held consists of an aggregate of (i) 8,750 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 8,750 shares of common stock and (ii) 11,199 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 11,199 shares of common stock. The address for Lynda Ludwig is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (46) | The number of shares of common stock held consists of 12,774 shares of Series B-1 Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 12,774 shares of common stock. The address for GNGI Investment Services LLLP is 5995 E Grant Rd, Suite 200, Tucson, AZ 85712. |
| (47) | The number of shares of common stock held in aggregate by Madison Trust Company, Custodian FBO Victor Estrada M1604015 and Victor A Estrada GST with Victor A Estrada as beneficiary consists of an aggregate of (i) 133,779 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 133,779 shares of common stock and (ii) 9,764 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 9,764 shares of common stock. The address for Victor A Estrada is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (48) | The number of shares of common stock held consists of 8,876 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 8,876 shares of common stock. The address for Jerry Collins is 15604 E. Chicory Dr., Fountain Hills, AZ 85268. |
| (49) | The number of shares of common stock held consists of an aggregate of (i) 16,906 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 16,906 shares of common stock and (ii) 8,506 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 8,506 shares of common stock. The address for Ivan I Smith III is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (50) | The number of shares of common stock held consists of 8,506 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 8,506 shares of common stock. The address for James Sage is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (51) | The number of shares of common stock held consists of 8,387 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 8,387 shares of common stock. The address for Chris McCall is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (52) | The number of shares of common stock held consists of 10,644 shares of Series B-1 Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 10,644 shares of common stock. The address for Gladys E. Pierson is 122 West Davis Street, Ste 110, Conroe, TX 77301. |
| (53) | The number of shares of common stock held consists of an aggregate of (i) 30,637 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 30,637 shares of common stock and (ii) 8,188 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 8,188 shares of common stock. The address for Schnoy, LLC is 7411 E. Calle Cabo, Tucson, AZ 85750. |
| (54) | The number of shares of common stock held consists of 8,136 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 8,136 shares of common stock. The address for Marshall A. Lewis is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (55) | The number of shares of common stock held consists of 7,692 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 7,692 shares of common stock. The address for John Mai is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (56) | The number of shares of common stock held consists of 7,316 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 7,316 shares of common stock. The address for Barbara Peraskevas is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (57) | The number of shares of common stock held consists of an aggregate of (i) 5,881 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 5,881 shares of common stock and (ii) 7,101 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 7,101 shares of common stock. The address for Sabio Viloria is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (58) | The number of shares of common stock held consists of 7,101 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 7,101 shares of common stock. The address for Chad Burns is 215 Magnolia Ridge Pl, Apt 206, Dothan, AL 36303. |
| (59) | The number of shares of common stock held in aggregate by Greg Maher and Vantage FBO Gregory Maher IRA with Gregory Maher as beneficiary consists of an aggregate of (i) 14,702 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 14,702 shares of common stock and (ii) 6,472 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 6,472 shares of common stock. The address for Greg Maher is 13020 N 82nd Street, Scottsdale, AZ 85260. |
| (60) | The number of shares of common stock held consists of 7,096 shares of Series B-1 Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 7,096 shares of common stock. The address for Robert J Anthony is 661 Bering Dr, Unit 602, Houston, TX 77057. |
| (61) | The number of shares of common stock held consists of 5,104 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 5,104 shares of common stock. The address for Steve Rice is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (62) | The number of shares of common stock held consists of 4,863 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,863 shares of common stock. The address for Rich Sangberg is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (63) | The number of shares of common stock held consists of 4,808 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,808 shares of common stock. The address for Charles Cadenhead is 2303 County Road 582A, Brazoria, TX 77422. |
| (64) | The number of shares of common stock held consists of 4,623 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,623 shares of common stock. The address for RoboSpine LLC is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (65) | The number of shares of common stock held in aggregate by Ted Fowler, 2Xd Productions 401K FBO Ted Fowler, and Vantage Retirement Ted Fowler with Ted Fowler as beneficiary consists of an aggregate of (i) 47,839 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 47,839 shares of common stock and (ii) 4,582 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,582 shares of common stock. The address for Ted Fowler is 2422 Iron Canyon Dr, Park City, UT 84060. |
| (66) | The number of shares of common stock held consists of an aggregate of (i) 25,455 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 25,455 shares of common stock and (ii) 4,438 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,438 shares of common stock. The address for J. Dickson Mappus is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (67) | The number of shares of common stock held consists of an aggregate of (i) 12,728 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 12,728 shares of common stock and (ii) 4,438 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,438 shares of common stock. The address for Mark Drinkwater is 7715 E. Montebello Ave, Scottsdale, AZ 85250. |
| (68) | The number of shares of common stock held consists of 4,438 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,438 shares of common stock. The address for Darrin Shandley is 1540 Wildcat Drive, Portland, TX 78374. |
| (69) | The number of shares of common stock held consists of 4,438 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,438 shares of common stock. The address for Mary Taormino is 615 Buchanan St, Davis, CA 95616. |
| (70) | The number of shares of common stock held consists of 4,068 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 4,068 shares of common stock. The address for Wichuta Prochnow is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (71) | The number of shares of common stock held consists of 3,938 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 3,938 shares of common stock. The address for Bart Pasternak is 1 Canfield Crossing Rd, Norwalk, CT 06855. |
| (72) | The number of shares of common stock held consists of 3,883 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 3,883 shares of common stock. The address for Charles Read is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (73) | The number of shares of common stock held consists of 3,872 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 3,872 shares of common stock. The address for Sheryl Moses is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (74) | The number of shares of common stock held consists of 3,750 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 3,750 shares of common stock. The address for Robert Myers Retirement Plan-Roth is 8390 E Via de Ventura Ste F110 #169, Scottsdale, AZ 85258. |
| (75) | The number of shares of common stock held consists of an aggregate of (i) 38,314 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 38,314 shares of common stock and (ii) 3,551 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 3,551 shares of common stock. The address for William Ford is c/o FibroBiologics, 455 E Medical Center Blvd, Ste 300, Houston, TX 77598. |
| (76) | The number of shares of common stock held consists of 1,480 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 1,480 shares of common stock. The address for Gloria E Lopez is 4330 N. Chapel Rd, Franklin, TN 37067. |
| (77) | The number of shares of common stock held consists of 740 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 740 shares of common stock. The address for Donna L Brown is 1292 Greenbrier Road, West Sacramento, CA 95691. |
| (78) | The number of shares of common stock held consists of 5,925 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 5,925 shares of common stock. The address for Jason D Sellers is PO Box 41504, Nashville, TN 37204. |
| (79) | The number of shares of common stock held by 675 shareholders consists of an aggregate of (i) 376,214 shares of non-voting common stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 376,214 shares of common stock, (ii) 222,554 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 222,554 shares of common stock, and (ii) 11,172 shares of Series B Preferred Stock which, in connection with the Direct Listing, will automatically convert, on a one-for-one basis, into 11,172 shares of common stock. The combined voting power of these shares to be registered in connection with the Direct Listing is less than 1% of the Total Voting Power. |
| 115 |
General
The following description summarizes certain important terms of our capital stock, as they are expected to be in effect in connection with the effectiveness of the registration statement of which this prospectus forms a part. We expect to adopt an amended and restated certificate of incorporation and an amended and restated bylaws that will become effective in connection with the effectiveness of the registration statement of which this prospectus forms a part, and this description summarizes the provisions that are expected to be included in such documents. Because it is only a summary, it does not contain all the information that may be important to you. For a complete description of the matters set forth in this section titled “Description of Capital Stock,” you should refer to our amended and restated certificate of incorporation and our amended and restated bylaws, which are included as exhibits to the registration statement of which this prospectus forms a part, and to the applicable provisions of Delaware law.
In connection with the Direct Listing, (i) all of our then outstanding Series A Preferred Stock, all of which are held by FibroGenesis, will be automatically canceled without the payment of additional consideration by or to the holder thereof, (ii) all of our then outstanding non-voting common stock will automatically convert, without the payment of additional consideration by or to the holder thereof, into common stock, on a one-for-one basis, (iii) all of our then outstanding Series B Preferred Stock and all of our then outstanding Series B-1 Preferred Stock will automatically convert, without the payment of additional consideration by or to the holder thereof, into common stock, on a one-for-one basis and (iv) all of our then outstanding Series C Preferred Stock will remain Series C Preferred Stock, such that, immediately after the Direct Listing, our issued and outstanding capital stock will consist of common stock and Series C Preferred Stock.
Upon consummation of the Direct Listing, after giving effect to the filing and effectiveness of our amended and restated certificate of incorporation and the adoption of our amended and restated bylaws, we will be authorized to issue 110,000,000 shares of capital stock, which will consist of: (i) 100,000,000 shares of common stock, par value $0.00001 per share and (ii) 10,000,000 shares of preferred stock, par value $0.00001 per share, of which 2,500 shares are designated as Series C Preferred Stock.
After giving effect to the Reverse Stock Split and the automatic conversion, in connection with the Direct Listing, of all of our then outstanding non-voting common stock and convertible preferred stock (being our Series B Preferred Stock and Series B-1 Preferred Stock), as of September 30, 2023, there were 32,477,209 shares of our common stock outstanding, held by 1,178 stockholders of record, and 2,500 shares of our Series C Preferred Stock, being all of the authorized Series C Preferred Stock, outstanding, held by one stockholder of record. Pursuant to our amended and restated certificate of incorporation, our board of directors will have the authority, without stockholder approval except as required by Nasdaq rules, to issue additional shares of our capital stock.
Common Stock
Our amended and restated certificate of incorporation will provide that:
| ● | holders of common stock will have voting rights for the election of our directors and all other matters requiring stockholder action, except with respect to amendments to our certificate of incorporation that alter or change the powers, preferences, rights or other terms of any outstanding preferred stock if the holders of such affected series of preferred stock are entitled to vote on such an amendment; | |
| ● | holders of common stock will be entitled to one vote per share on matters to be voted on by stockholders and also will be entitled to receive such dividends, if any, as may be declared from time to time by our board of directors in its discretion out of funds legally available therefor; | |
| ● | the payment of dividends, if any, on the common stock will be subject to the prior payment of dividends on any outstanding preferred stock; | |
| ● | upon our liquidation or dissolution, the holders of common stock will be entitled to receive pro rata all assets remaining available for distribution to stockholders after payment of all liabilities and provision for the liquidation of any shares of preferred stock outstanding at that time; and | |
| ● | our stockholders have no conversion, preemptive or other subscription rights and there are no sinking fund or redemption provisions applicable to the common stock. |
Preferred Stock
Our amended and restated certificate of incorporation will provide that shares of preferred stock may be issued from time to time in one or more series. Our board of directors will be authorized to fix the voting rights, if any, designations, powers, preferences, the relative, participating, optional or other special rights, if any, and any qualifications, limitations and restrictions thereof, applicable to the shares of each series. Our board of directors will be able to, without stockholder approval, issue preferred stock with voting and other rights that could adversely affect the voting power and other rights of the holders of the common stock and could have anti-takeover effects. The ability of our board of directors to issue preferred stock without stockholder approval could have the effect of delaying, deferring or preventing a change of our control or the removal of our existing management.
| 116 |
Series C Preferred Stock
Upon consummation of the Direct Listing, there will be one series of designated preferred stock, being the Series C Preferred Stock, 2,500 total shares of which are authorized and all of which 2,500 authorized shares of Series C Preferred Stock will be issued, outstanding and held by Pete O’Heeron, our founder, Chief Executive Officer and Chairperson of our board of directors. The outstanding shares of Series C Preferred Stock are fully paid and nonassessable.
The Series C Preferred Stock shall rank senior to our common stock upon our liquidation, dissolution, winding up or otherwise.
Upon consummation of the Direct Listing, the Series C Preferred Stock shall be entitled to vote on any matter to be voted on by our stockholders, in each case voting together with the holders of our common stock as a single class, and each share of Series C Preferred Stock shall be entitled to 13,000 votes. The Series C Preferred Stock shall be entitled to receive the same prior notice of any meeting of stockholders as provided to our common stockholders.
The Series C Preferred Stock shall not be entitled to any dividend, whether payable in cash, stock or property.
Subject to the superior rights of other, then outstanding, classes or series of preferred stock, in the event of any liquidation, dissolution or winding up of our company, the Series C Preferred Stock shall be entitled to receive, prior and in preference to any distribution in such liquidation, dissolution or winding up of any of our assets to the holders of our common stock, a liquidation preference of $18.00 per share (subject to appropriate adjustment in the event of any stock split, combination or other similar recapitalization).
The Series C Preferred Stock may be converted at any time as follows:
| ● | At the option of the holder, a share of Series C Preferred Stock may be converted into one share of our common stock; and | |
| ● | Upon the election of the holders of a majority of the then outstanding shares of Series C Preferred Stock, all outstanding shares of Series C Preferred Stock may be converted into an equal number of shares of our common stock, on a one-for-one basis. |
In addition, the Series C Preferred Stock is subject to a mandatory conversion upon any transfer of the Series C Preferred Stock. Each share of Series C Preferred Stock shall automatically convert, without the payment of additional consideration by or to the holder thereof, into one fully paid and non-assessable share of our common stock, upon any transfer of any share of Series C Preferred Stock, whether or not for value. Any shares of Series C Preferred Stock converted as described above must be retired and cancelled and may not be reissued as shares of such series.
For as long as the Series C Preferred Stock remain outstanding, the aggregate number of shares of Series C Preferred Stock then outstanding, shall be proportionately adjusted for any increase or decrease in the number of issued shares of our common Stock resulting from a subdivision or combination of our common stock or other similar recapitalization, in each case effected without our receipt of consideration.
The Series C Preferred Stock will be subject to an irrevocable proxy issued by Pete O’Heeron, the holder of all of the Series C Preferred Stock, in favor and for the benefit of, our board of directors, granting our board of directors the irrevocable proxy, for as long as the Series C Preferred Stock remain outstanding, to vote all of the Series C Preferred Stock on all matters on which the Series C Preferred Stock are entitled to vote, in any manner that our board of directors may determine in its sole and absolute discretion; provided, however, that such irrevocable proxy shall not, without the written consent of Pete O’Heeron, permit our board of directors to vote the Series Preferred Stock with respect to any proposal to amend, delete or waive any rights of Pete O’Heeron with respect to the Series C Preferred Stock as set forth in our amended and restated certificate of incorporation. In light of the superior voting rights associated with the Series C Preferred Stock, the irrevocable proxy is intended to ensure that such superior voting rights are utilized in our best interest and to avoid or mitigate conflicts that may arise in the future for Pete O’Heeron as an individual stockholder employee.
| 117 |
Anti-Takeover Effects of our Certificate of Incorporation, Bylaws and Delaware Law
Our amended and restated certificate of incorporation and amended and restated bylaws will include a number of provisions that may have the effect of delaying, deferring or preventing another party from acquiring control of us and encouraging persons considering unsolicited tender offers or other unilateral takeover proposals to negotiate with our board of directors rather than pursue non-negotiated takeover attempts. These provisions include the items described below.
Classified Board
Our amended and restated certificate of incorporation will require our board of directors to be divided into three classes serving staggered three-year terms, with one class elected each year. The classification of directors has the effect of making it more difficult for stockholders to change the composition of our board of directors.
Stockholder Actions by Written Consent
Our amended and restated certificate of incorporation will require that, any action required or permitted to be taken by our stockholders must be effected at a duly-called annual or special meeting of our stockholders and may not be effected by written consent in lieu of a meeting.
Advance Notice Requirements
Our amended and restated bylaws will establish advance notice procedures with regard to stockholder proposals relating to the nomination of candidates for election as directors or new business to be brought before meetings of our stockholders. These procedures will specify that notice of stockholder proposals must be timely given in writing to our corporate secretary prior to the meeting at which the action is to be taken, and define what is considered timely. Our amended and restated bylaws will also specify the requirements as to form and content of all stockholder notices. These requirements may preclude stockholders from bringing matters before the stockholders at an annual or special meeting.
Director Removal and Vacancies
Our amended and restated certificate of incorporation will require that, a member of our board of directors or our entire board may only be removed for cause, and then only by the affirmative vote of the holders of at least 662/3% in voting power of our stock entitled to vote on such removal. In addition, our amended and restated certificate of incorporation will require that, any newly created directorship that results from an increase in the number of directors or any vacancy on our board of directors, must be filled solely by the affirmative vote of a majority of the total number of directors then in office, even if less than a quorum, or by a sole remaining director and may not be filled by the stockholders.
Supermajority Voting Requirements
Our amended and restated certificate of incorporation will require the affirmative vote of the holders of at least 662/3% in voting power of our stock entitled to vote thereon to (i) amend, alter or repeal our bylaws and adopt new bylaws or (ii) to amend, alter, change or repeal, or adopt any provision inconsistent with, certain provisions of our certificate of incorporation, including the provisions relating to the requirement to have a classified board, the provisions relating to the removal of directors, the provision precluding stockholder action by written consent and the choice of forum provision in our amended and restated certificate of incorporation (as explained below).
Undesignated Preferred Stock
Our amended and restated certificate of incorporation will provide for authorized shares of preferred stock. The existence of authorized but unissued shares of preferred stock may enable our board of directors to discourage an attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise. For example, if in the due exercise of its fiduciary obligations, our board of directors were to determine that a takeover proposal is not in the best interests of our shareholders, our board of directors could cause shares of preferred stock to be issued without shareholder approval in one or more private offerings or other transactions that might dilute the voting or other rights of the proposed acquirer or insurgent shareholder or shareholder group. In this regard, our amended and restated certificate of incorporation will grant our board of directors broad power to establish the rights and preferences of authorized and unissued shares of preferred stock. The issuance of shares of preferred stock could decrease the amount of earnings and assets available for distribution to holders of shares of common stock. The issuance may also adversely affect the rights and powers, including voting rights, of these holders and may have the effect of delaying, deterring or preventing a change in our control.
| 118 |
Exclusive Forum
Our amended and restated certificate of incorporation will provide that, unless we consent in writing to the selection of an alternative forum, the (i) Court of Chancery of the State of Delaware (or, if the Court of Chancery does not have jurisdiction, the federal district court for the District of Delaware) shall, to the fullest extent permitted by law, be the sole and exclusive forum for (a) any derivative action or proceeding brought on our behalf, (b) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees to us or our stockholders, (c) any action arising pursuant to any provision of the DGCL, our certificate of incorporation or our bylaws or (d) any action asserting a claim governed by the internal affairs doctrine and (ii) to the fullest extent permitted by law, the federal district courts of the United States of America shall be the exclusive forum for the resolution of any complaint asserting a cause or causes of action arising under the Securities Act, including all causes of action asserted against any defendant to such complaint. The foregoing provision would not preclude stockholders that assert claims under the Exchange Act from bringing such claims in federal court, to the extent that the Exchange Act confers exclusive federal jurisdiction over such claims, subject to applicable law. Our choice of forum provision may impose additional litigation costs on stockholders in pursuing claims and may limit a stockholder’s ability to bring a claim in a judicial forum that it believes to be favorable for disputes with us or any of our directors, officers or other employees, which may discourage lawsuits with respect to such claims.
Limitation of Liability and Indemnification of Directors and Officers
Our amended and restated bylaws will provide that our directors and officers will be indemnified by us to the fullest extent authorized by Delaware law.
These provisions may discourage stockholders from bringing a lawsuit against our directors for breach of their fiduciary duty. These provisions also may have the effect of reducing the likelihood of derivative litigation against directors and officers, even though such an action, if successful, might otherwise benefit us and our stockholders. Furthermore, a stockholder’s investment may be adversely affected to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions. We believe that these provisions and insurance are necessary to attract and retain talented and experienced directors and officers. In addition, in connection with the effectiveness of the registration statement of which this prospectus forms a part, we intend to enter into separate indemnification agreements with each of our directors and executive officers.
Section 203 of the DGCL
As a Delaware corporation, we will be subject to the provisions of Section 203 of the DGCL. This statute prevents certain Delaware corporations, under certain circumstances, from engaging in a “business combination” with an “interested stockholder.” In general, Section 203 defines an “interested stockholder” as an entity or person who, together with the person’s affiliates and associates, beneficially owns 15% or more of the outstanding voting stock of the corporation.
A “business combination” includes a merger or sale of more than 10% of our assets. However, the above provisions of Section 203 of the DGCL do not apply if:
| ● | the business combination takes place more than three years after the interested stockholder became an “interested stockholder;” | |
| ● | our board of directors approves the transaction that made the stockholder an “interested stockholder” prior to the date of the transaction; | |
| ● | after the completion of the transaction that resulted in the stockholder becoming an interested stockholder, that stockholder owned at least 85% of our voting stock outstanding, other than statutorily excluded shares of common stock; or | |
| ● | on or subsequent to the date of the transaction, the business combination is approved by our board of directors and authorized at a meeting of our stockholders, and not by written consent, by an affirmative vote of at least two-thirds of the outstanding voting stock not owned by the interested stockholder. |
Listing
We have applied to list our common stock on the Nasdaq Global Market under the symbol “FBLG”.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is VStock Transfer LLC. The transfer agent and registrar’s address is 18 Lafayette Place, Woodmere, NY 11598. The transfer agent and registrar can be contacted by phone at: (212) 828-8436.
| 119 |
SHARES ELIGIBLE FOR FUTURE SALE
Prior to the listing of our common stock on Nasdaq, there has been no public market for our common stock. Sales of a substantial number of shares our common stock in the public market following our listing on Nasdaq, or the perception that such sales could occur, could adversely affect the public price of our common stock and may make it more difficult for you to sell your shares at a time and price that you deem appropriate. We will have no input if and when any Registered Stockholders may, or may not, elect to sell their shares or the prices at which any such sales may occur.
After the Direct Listing, a total of 32,492,068 shares of our common stock will be outstanding, including 4,806,226 shares of our common stock registered for resale under the registration statement of which this prospectus forms a part. Any shares not registered hereunder will be “restricted securities,” as that term is defined in Rule 144 under the Securities Act. These restricted securities are eligible for public sale only if they are registered under the Securities Act, including, but not limited to, the shares registered hereunder, or if they qualify for an exemption from registration, including under Rules 144 or 701 under the Securities Act, which are summarized below. Restricted securities also may be sold outside of the United States to non-U.S. persons in accordance with Rule 904 of Regulation S. With the exception of shares owned by our directors, officers and certain stockholders, substantially all of our common stock may be sold after our initial listing on Nasdaq, either by the Registered Stockholders pursuant to this prospectus or by our other existing stockholders in accordance with Rule 144 of the Securities Act.
Rule 144
In general, under Rule 144 as currently in effect, once we have been subject to and in compliance with public company reporting requirements of Section 13 or Section 15(d) of the Exchange Act for at least 90 days, an eligible shareholder is entitled to sell such shares without complying with the manner of sale, volume limitation, or notice provisions of Rule 144, subject to compliance with the public information requirements of Rule 144. To be an eligible shareholder under Rule 144, such shareholder must not be deemed to have been one of our affiliates for purposes of the Securities Act at any time during the 90 days preceding a sale and who has beneficially owned the shares of common stock proposed to be sold for at least six months, including the holding period of any prior owner other than our affiliates. If such a person has beneficially owned the shares of common stock proposed to be sold for at least one year, including the holding period of any prior owner other than our affiliates, then such person is entitled to sell such shares without complying with any of the requirements of Rule 144.
In general, under Rule 144, as currently in effect, our affiliates or persons selling common stock on behalf of our affiliates are entitled to sell shares 90 days after we become a reporting company. Within any three-month period, such shareholders may sell a number of shares that does not exceed the greater of:
| ● | 1% of the number of shares of common stock then outstanding, which will equal approximately shares immediately after our registration; or | |
| ● | the average weekly trading volume of our common stock during the four calendar weeks preceding the filing of a notice on Form 144 with respect to such sale. |
Sales under Rule 144 by our affiliates or persons selling shares of common stock on behalf of our affiliates also are subject to certain manner of sale provisions and notice requirements and to the availability of current public information about us.
Rule 701
Rule 701 generally allows a shareholder who was issued shares under a written compensatory plan or contract and who is not deemed to have been our affiliate during the immediately preceding 90 days, to sell these shares in reliance on Rule 144, but without being required to comply with the public information, holding period, volume limitation, or notice provisions of Rule 144. Rule 701 also permits our affiliates to sell their Rule 701 shares under Rule 144 without complying with the holding period requirements of Rule 144. All holders of Rule 701 shares, however, are required by that rule to wait until 90 days after we become a reporting company before selling those shares under Rule 701.
Registration Statements on Form S-8
We intend to file one or more registration statements on Form S-8 under the Securities Act to register shares of our common stock subject to outstanding stock options or reserved for issuance under our 2022 Stock Plan, as soon as permitted under the Securities Act. Such registration statements will automatically become effective upon filing with the SEC. However, shares registered on Form S-8 may be subject to the volume limitations and the manner of sale, notice, and public information requirements of Rule 144.
| 120 |
SALE PRICE HISTORY OF OUR CAPITAL STOCK
We have applied to list our common stock on Nasdaq. Prior to the listing of our common stock on Nasdaq, there has been no public market for our common stock. Our common stock has a limited history of trading in private transactions. In December 2022, we issued an aggregate of the equivalent of 381,658 shares of Series B Preferred Stock to investors in a private placement, at a price the equivalent of $6.76 per share as to the equivalent of 318,049 shares, with the remaining equivalent of 63,609 shares being bonus shares. The bonus shares were issued to investors that sent payment within one week of signing the subscription agreement. From February 2023 through April 2023, we issued an aggregate of the equivalent of 890,310 shares of Series B Preferred Stock to investors in a Regulation Crowdfunding offering, at a price the equivalent of $6.76 per share as to the equivalent of 724,937 shares, with the remaining equivalent of 143,225 shares and equivalent of 22,148 shares being bonus shares and commission payment shares, respectively. The bonus shares were issued in accordance with the offering to investors that met either one, or a combination, of loyalty, early-bird timing, amount-based, and/or StartEngine owners’ bonus requirements. Existing shareholders of FibroBiologics who invested in the Regulation Crowdfunding offering received loyalty 10% bonus shares. Investors who invested at least $1,000 within the first two weeks of the Regulation Crowdfunding offering received timing-based 5% bonus shares. Investors who invested at least $25,000 within the first 72 hours of the Regulation Crowdfunding offering received timing-based 10% bonus shares. Investors who invested at least $25,000 in the Regulation Crowdfunding offering received amount-based 5% bonus shares. Investors who invested at least $100,000 in the Regulation Crowdfunding offering received amount-based 10% bonus shares. Investors who were eligible for the StartEngine Crowdfunding Inc. OWNer’s bonus received 10% bonus shares. Investors in the Regulation Crowdfunding offering received the highest amount-based or timing-based bonus for which they were eligible and the loyalty bonus and OWNer’s bonus for which they were eligible. In March and April 2023, we issued the equivalent of 1,680,084 shares of Series B Preferred Stock to investors in private placements, at a price the equivalent of $6.76 per share as to the equivalent of 1,527,349 shares, with the remaining equivalent of 152,735 shares being bonus shares. The bonus shares were issued to investors that sent payment within one week of signing the subscription agreement. From April 2023 through September 2023, we issued in a private placement the equivalent of 74,922 shares of Series B-1 Preferred Stock to investors in a private placement, at prices ranging from the equivalent of $18.00 to $20.00 per share as to the equivalent of 64,070 shares, with the remaining equivalent of 10,852 shares being bonus shares. The bonus shares were issued to investors that sent payment within one week of signing the subscription agreement. In connection with a portion of such private placement of our Series B-1 Preferred Stock, we also agreed to issue warrants, exercisable for a period of three years from our Direct Listing, to purchase an aggregate of the equivalent of an aggregate of 8,890 shares of our common stock at an exercise price of the equivalent of $20.00 per share. In November 2023, the Company issued a total of 14,859 additional shares of Series B-1 Preferred Stock and 1,431 additional warrants to purchase shares of common stock to investors who subscribed to purchase shares of Series B-1 Preferred Stock at a price per share that exceeded the reference price per share expected in the Direct Listing. While the Advisor is expected to consider this information in connection with setting the opening public price of our common stock, this information may have little or no relation to broader market demand for our common stock and thus the opening public price and subsequent public price of our common stock on Nasdaq. As a result, you should not place undue reliance on these historical private sale prices as it may differ materially from the opening public price and subsequent public price of our common stock on Nasdaq.
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS
The following is a general discussion of material U.S. federal income tax considerations and certain U.S. federal estate tax considerations relating to the acquisition, ownership, and disposition of our common stock applicable to non-U.S. holders that purchase our common stock in this offering and hold it as a “capital asset” within the meaning of Section 1221 of the U.S. Internal Revenue Code of 1986, as amended, or the Code (generally, property held for investment). For purposes of this discussion, a “non-U.S. holder” means a beneficial owner of our common stock (other than an entity or arrangement that is treated as a partnership for U.S. federal income tax purposes) that is not, for U.S. federal income tax purposes, any of the following:
| ● | an individual who is a citizen or resident of the United States; | |
| ● | a corporation (or entity treated as a corporation for United States federal income tax purposes) created or organized in or under the laws of the United States, any state thereof or the District of Columbia; | |
| ● | an estate, the income of which is includable in gross income for U.S. federal income tax purposes regardless of its source; or | |
| ● | a trust if (i) a court within the United States is able to exercise primary supervision over the administration of the trust and one or more “United States persons,” as defined under the Code, or U.S. persons, have the authority to control all substantial decisions of the trust or (ii) such trust has made a valid election to be treated as a U.S. person for U.S. federal income tax purposes. |
If a partnership (or other entity or arrangement treated as a partnership for U.S. federal income tax purposes) holds our common stock, the tax treatment of a partner therein will generally depend on the status of the partner and the activities of the partnership. Partners of a partnership holding our common stock should consult their tax advisors as to the particular U.S. federal income tax consequences applicable to them.
This discussion is based on current provisions of the Code, final, temporary and proposed Treasury regulations promulgated thereunder, or the Treasury Regulations, judicial decisions, published rulings and administrative pronouncements of the U.S. Internal Revenue Service, or IRS, all as in effect as of the date of this prospectus and all of which are subject to change or to differing interpretation, possibly with retroactive effect. Any change could alter the tax consequences to non-U.S. holders described herein. There can be no assurance that the IRS will not challenge one or more of the tax consequences described herein.
| 121 |
This discussion does not address all aspects of U.S. federal income and estate taxation that may be relevant to a particular non-U.S. holder in light of that non-U.S. holder’s individual circumstances nor does it address any aspects of U.S. state, local or non-U.S. taxes, other U.S. federal tax, the alternative minimum tax, or the unearned income Medicare contribution tax on net investment income. This discussion also does not consider any specific facts or circumstances that may apply to a non-U.S. holder and does not address the special tax rules applicable to particular non-U.S. holders, such as:
| ● | banks, insurance companies and other financial institutions; | |
| ● | brokers or dealers or traders in securities; | |
| ● | tax-exempt organizations; | |
| ● | pension plans; | |
| ● | persons who hold our common stock as part of a straddle, hedge, conversion transaction, synthetic security or other integrated investment or who have elected to mark securities to market; | |
| ● | controlled foreign corporations, passive foreign investment companies, and corporations that accumulate earnings to avoid U.S. federal income tax; | |
| ● | non-U.S. governments; and | |
| ● | U.S. expatriates and former citizens or long-term residents of the United States. |
| 122 |
THIS SUMMARY IS NOT INTENDED TO CONSTITUTE A COMPLETE DESCRIPTION OF ALL TAX CONSEQUENCES FOR NON-U.S. HOLDERS RELATING TO THE OWNERSHIP AND DISPOSITION OF OUR COMMON STOCK. PROSPECTIVE HOLDERS OF OUR COMMON STOCK SHOULD CONSULT WITH THEIR TAX ADVISORS REGARDING THE TAX CONSEQUENCES TO THEM (INCLUDING THE APPLICATION AND EFFECT OF ANY STATE, LOCAL, NON-U.S. INCOME AND OTHER TAX LAWS) OF THE ACQUISITION, OWNERSHIP AND DISPOSITION OF OUR COMMON STOCK.
Distributions
As discussed under “Dividend Policy” above, we do not expect to make distributions on our common stock in the foreseeable future. However, if we do make distributions of cash or property on our common stock, such distributions will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Amounts of distributions not treated as dividends for U.S. federal income tax purposes will first constitute a tax-free return of capital of the non-U.S. holder’s investment and be applied against and reduce a non-U.S. holder’s adjusted tax basis in its common stock, but not below zero. Any remaining excess will be treated as capital gain and will be treated as described below under “Gain on Sale or Other Disposition of Common Stock.” Because we may not know the extent to which a distribution is a dividend for U.S. federal income tax purposes at the time it is made, for purposes of the withholding rules discussed below we or the applicable withholding agent may treat the entire distribution as a dividend. Any such distributions will also be subject to the discussions below under the headings “FATCA” and “Backup Withholding, Information Reporting and Other Reporting Requirements.”
Subject to the discussion in the next two paragraphs, dividends paid to a non-U.S. holder generally will be subject to withholding of U.S. federal income tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty between the United States and such holder’s country of residence.
Dividends we pay to a non-U.S. holder that are effectively connected with such non-U.S. holder’s conduct of a trade or business within the United States (and, if required by an applicable tax treaty, are attributable to a U.S. permanent establishment or a fixed base maintained by such non-U.S. holder) will generally be exempt from the U.S. federal withholding tax described above, if the non-U.S. holder complies with applicable certification and disclosure requirements (generally including provision of a valid IRS Form W-8ECI (or applicable successor form) certifying that the dividends are effectively connected with the non-U.S. holder’s conduct of a trade or business within the United States). Instead, such dividends generally will be subject to U.S. federal income tax on a net income basis, at regular U.S. federal income tax rates as would apply if such holder were a U.S. person (as defined in the Code). Any U.S. effectively connected income received by a non-U.S. holder that is classified as a corporation for U.S. federal income tax purposes may also be subject to an additional “branch profits tax” at a rate of 30% (or such lower rate as may be specified by an applicable income tax treaty).
| 123 |
A non-U.S. holder of our common stock who claims the benefit of an applicable income tax treaty between the United States and such holder’s country of residence generally will be required to provide a properly executed IRS Form W-8BEN or W-8BEN-E (or successor form) and satisfy applicable certification and other requirements. Non-U.S. holders are urged to consult their tax advisors regarding their entitlement to benefits under a relevant income tax treaty and the specific methods available to them to satisfy these requirements.
Gain on Sale or Other Disposition of Common Stock
Subject to the discussion below under the headings “FATCA” and “Backup Withholding, Information Reporting and Other Reporting Requirements,” a non-U.S. holder generally will not be subject to U.S. federal income tax on any gain realized upon the sale or other disposition of the non-U.S. holder’s shares of our common stock unless:
| ● | the gain is effectively connected with a trade or business carried on by the non-U.S. holder within the United States (and, if required by an applicable income tax treaty, is attributable to a U.S. permanent establishment or fixed base maintained by such non-U.S. holder); | |
| ● | the non-U.S. holder is an individual and is present in the United States for 183 days or more in the taxable year of disposition and certain other conditions are met; or | |
| ● | we are or have been a “U.S. real property holding corporation” for U.S. federal income tax purposes at any time within the shorter of the five-year period preceding such disposition or such non-U.S. holder’s holding period of our common stock, and, provided that our common stock is regularly traded in an established securities market within the meaning of applicable Treasury Regulations, the non-U.S. holder has held, directly, indirectly, or constructively, at any time during said period, more than 5% of our common stock. |
Gain that is effectively connected with the conduct of a trade or business in the United States generally will be subject to U.S. federal income tax on a net income tax basis, at regular U.S. federal income tax rates that apply to U.S. persons. If the non-U.S. holder is a non-U.S. corporation, the branch profits tax described above also may apply to such effectively connected gain. An individual non-U.S. holder who is subject to U.S. federal income tax because the non-U.S. holder was present in the United States for 183 days or more during the year of sale or other disposition of our common stock will be subject to a flat 30% tax (or such lower rate as may be specified by an applicable income tax treaty) on the gain derived from such sale or other disposition, which may be offset by certain U.S. source capital losses, if any. We believe that we are not and we do not anticipate becoming a U.S. real property holding corporation for U.S. federal income tax purposes. Non-U.S. holders should consult their tax advisors regarding potentially applicable income tax treaties that may provide for different rules.
FATCA
Withholding taxes may be imposed under the Foreign Account Tax Compliance Act, or FATCA, on certain types of payments made to non-U.S. financial institutions and certain other non-U.S. entities. Specifically, a 30% withholding tax may be imposed on dividends (including deemed dividends) paid on our common stock, to a “foreign financial institution” or a “non-financial foreign entity” (each as defined in the Code), unless (i) the foreign financial institution undertakes certain diligence and reporting obligations, (ii) the non-financial foreign entity either certifies it does not have any “substantial United States owners” (as defined in the Code) or furnishes identifying information regarding each substantial U.S. owner, or (iii) the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from these rules. Foreign financial institutions located in jurisdictions that have an intergovernmental agreement with the U.S. governing FATCA may be subject to the reporting rules of that intergovernmental agreement. Because we may not know the extent to which a distribution is a dividend for U.S. federal income tax purposes at the time it is made, for purposes of these withholding rules we or the applicable withholding agent may treat the entire distribution as a dividend. Although withholding under FATCA would have applied also to payments of gross proceeds from the sale or other disposition of stock on or after January 1, 2019, proposed Treasury Regulations would eliminate FATCA withholding on payments of gross proceeds entirely. Taxpayers generally may rely on these proposed Treasury Regulations until final Treasury Regulations are issued. Under certain circumstances, a non-U.S. holder will be eligible for refunds or credits of withholding taxes imposed under FATCA by timely filing a U.S. federal income tax return. Prospective investors should consult their tax advisors regarding the potential application of these withholding provisions.
| 124 |
Backup Withholding, Information Reporting and Other Reporting Requirements
We must report annually to the IRS and to each non-U.S. holder the amount of any distributions paid to, and the tax withheld with respect to, each non-U.S. holder. These reporting requirements apply regardless of whether withholding was reduced or eliminated by an applicable income tax treaty. Copies of this information reporting may also be made available under the provisions of a specific income tax treaty or agreement with the tax authorities in the country in which the non-U.S. holder resides or is established.
A non-U.S. holder will generally be subject to backup withholding for dividends on our common stock paid to such holder unless such holder certifies under penalties of perjury that, among other things, it is a non-U.S. holder (provided that the payor does not have actual knowledge or reason to know that such holder is a U.S. person) or otherwise establishes an exemption.
Information reporting and backup withholding generally will apply to the proceeds of a disposition of our common stock by a non-U.S. holder effected by or through the U.S. office of any broker, U.S. or non-U.S., unless the holder certifies its status as a non-U.S. holder and satisfies certain other requirements, or otherwise establishes an exemption. Generally, information reporting and backup withholding will not apply to a payment of disposition proceeds to a non-U.S. holder where the transaction is effected outside the United States through a non-U.S. office of a broker. However, for information reporting purposes, dispositions effected through a non-U.S. office of a broker with substantial U.S. ownership or operations generally will be treated in a manner similar to dispositions effected through a U.S. office of a broker. Non-U.S. holders should consult their tax advisors regarding the application of the information reporting and backup withholding rules to them.
Backup withholding is not an additional income tax. Any amounts withheld under the backup withholding rules from a payment to a non-U.S. holder generally can be credited against the non-U.S. holder’s U.S. federal income tax liability, if any, or refunded, provided that the required information is furnished to the IRS in a timely manner. Non-U.S. holders should consult their tax advisors regarding the application of the information reporting and backup withholding rules to them.
U.S. Federal Estate Tax
Shares of our common stock that are owned or treated as owned by an individual who is not a citizen or resident of the United States (as specially defined for U.S. federal estate tax purposes) at the time of death are considered U.S. situs assets and will be included in the individual’s gross estate for U.S. federal estate tax purposes. Such shares, therefore, may be subject to U.S. federal estate tax, unless an applicable estate tax or other treaty provides otherwise.
The preceding discussion of material U.S. federal income tax considerations and certain U.S. federal estate tax considerations is for information only. It is not legal or tax advice. Prospective investors should consult their tax advisors regarding the particular U.S. federal, state, local and non-U.S. tax consequences of acquiring, owning and disposing of our common stock, including the consequences of any proposed changes in applicable laws.
| 125 |
The Registered Stockholders, and their pledgees, donees, transferees, assignees, or other successors in interest may sell their shares of common stock covered hereby pursuant to brokerage transactions on Nasdaq, or other public exchanges or registered alternative trading venues, at prevailing market prices at any time after the common stock are listed for trading. We are not party to any arrangement with any Registered Stockholder or any broker-dealer with respect to sales of shares of common stock by the Registered Stockholders, except we have engaged a financial advisor with respect to certain other matters relating to the registration of our common stock and listing of our common stock, as further described below. As such, we do not anticipate receiving notice as to if and when any Registered Stockholder may, or may not, elect to sell their shares of common stock or the prices at which any such sales may occur, and there can be no assurance that any Registered Stockholders will sell any or all of their shares of common stock covered by this prospectus.
We will not receive any proceeds from the sale of shares of common stock by the Registered Stockholders. We will recognize costs related to this direct listing and our transition to a publicly-traded company consisting of professional fees and other expenses. We will expense these amounts in the period incurred and not deduct these costs from net proceeds to the issuer as they would be in an initial public offering.
On the day that our shares of common stock are initially listed on Nasdaq, Nasdaq will begin accepting, but not executing, pre-opening buy and sell orders and will begin to continuously generate the indicative Current Reference Price on the basis of such accepted orders. The Current Reference Price is calculated each second and, during a 10-minute “Display Only” period, is disseminated, along with other indicative imbalance information, to market participants by Nasdaq on its NOII and BookViewer tools. Following the “Display Only” period, a “Pre-Launch” period begins, during which the Advisor, in its capacity as our financial advisor, must notify Nasdaq that our shares are “ready to trade.” Once the Advisor has notified Nasdaq that our shares of common stock are ready to trade, Nasdaq will confirm the Current Reference Price for our shares of common stock, in accordance with Nasdaq rules. If the Advisor then approves proceeding at the Current Reference Price, the applicable orders that have been entered will then be executed at such price and regular trading of our shares of common stock on Nasdaq will commence, subject to Nasdaq conducting validation checks in accordance with Nasdaq rules.
Under Nasdaq rules, the Current Reference Price means: (i) the single price at which the maximum number of orders to buy or sell can be matched; (ii) if there is more than one price at which the maximum number of orders to buy or sell can be matched, then it is the price that minimizes the imbalance between orders to buy or sell (i.e. minimizes the number of shares that would remain unmatched at such price); (iii) if more than one price exists under (ii), then it is the entered price (i.e. the specified price entered in an order by a customer to buy or sell) at which our shares of common stock will remain unmatched (i.e. will not be bought or sold); and (iv) if more than one price exists under (iii), a price determined by Nasdaq in consultation with the Advisor in its capacity as our financial advisor. In the event that more than one price exists under (iii), the Advisor will exercise any consultation rights only to the extent that it can do so consistent with the anti-manipulation provisions of the federal securities laws, including Regulation M, or applicable relief granted thereunder.
In determining the Current Reference Price, Nasdaq’s cross algorithms will match orders that have been entered into and accepted by Nasdaq’s system. This occurs with respect to a potential Current Reference Price when orders to buy shares of common stock at an entered bid price that is greater than or equal to such potential Current Reference Price are matched with orders to sell a like number of shares of common stock at an entered asking price that is less than or equal to such potential Current Reference Price. To illustrate, as a hypothetical example of the calculation of the Current Reference Price, if Nasdaq’s cross algorithms matched all accepted orders as described above, and two limit orders remained — a limit order to buy 500 shares of common stock at an entered bid price of $10.01 per share and a limit order to sell 200 shares of common stock at an entered asking price of $10.00 per share — the Current Reference Price would be selected as follows:
| ● | Under clause (i), if the Current Reference Price is $10.00, then the maximum number of additional shares that can be matched is 200. If the Current Reference Price is $10.01, then the maximum number of additional shares that can be matched is also 200, which means that the same maximum number of additional shares would be matched at the price of either $10.00 or $10.01. |
| 126 |
| ● | Because more than one price under clause (i) exists, under clause (ii), the Current Reference Price would be the price that minimizes the imbalance between orders to buy or sell (i.e., minimizes the number of shares that would remain unmatched at such price). Selecting either $10.00 or $10.01 as the Current Reference Price would create the same imbalance in the limit orders that cannot be matched, because at either price 300 shares would not be matched. | |
| ● | Because more than one price under clause (ii) exists, under clause (iii), the Current Reference Price would be the entered price at which orders for shares of common stock at such entered price will remain unmatched. In such case, choosing $10.01 would cause 300 shares of the 500-share limit order with the entered price of $10.01 to remain unmatched, compared to choosing $10.00, where all 200 shares of the limit order with the entered price of $10.00 would be matched, and no shares at such entered price remain unmatched. Thus, Nasdaq would select $10.01 as the Current Reference Price, because orders for shares at such entered price will remain unmatched. The above example (including the prices) is provided solely by way of illustration. |
The Advisor will determine when our shares of common stock are ready to trade and approve proceeding at the Current Reference Price primarily based on considerations of volume, timing and price. In particular, the Advisor will determine, based primarily on pre-opening buy and sell orders, when a reasonable amount of volume will cross on the opening trade such that sufficient price discovery has been made to open trading at the Current Reference Price. If the Advisor does not approve proceeding at the Current Reference Price (for example, due to the absence of adequate pre-opening buy and sell interest), the Advisor will request that Nasdaq delay the opening until such a time that sufficient price discovery has been made to ensure that a reasonable amount of volume crosses on the opening trade. Further, in the highly unlikely event that Nasdaq consults with the Advisor as described in clause (iv) of the definition of Current Reference Price, the Advisor would request that Nasdaq delay the opening to ensure a single opening price within clauses (i), (ii) or (iii) of the definition of the Current Reference Price. Under Nasdaq rules, in the event of such delay, prior to terminating such delay, there will be a 10-minute “Display Only” period during which market participants may enter quotes and orders in shares of our common stock in Nasdaq systems. In addition, beginning at 4:00 a.m., market participants may enter orders in shares of our common stock on Nasdaq. Such orders will be accepted and entered into the system. After the conclusion of the 10-minute “Display Only” period, our common stock will enter a “Pre-Launch” period of indeterminate duration. The “Pre-Launch” period will end and shares of our common stock will be released for trading by Nasdaq when certain conditions are met, including Nasdaq’s receipt of notice from the Advisor that our shares of common stock are ready to trade, after which the Nasdaq system will calculate the Current Reference Price at that time and display it to the Advisor. If the Advisor then approves proceeding, the Nasdaq system will conduct certain validation checks. The Advisor, with concurrence of Nasdaq, may determine at any point during the delay process up through the conclusion of the “Pre-Launch” period to postpone and reschedule the Direct Listing. The Registered Stockholders will not be involved in Nasdaq’s price-setting mechanism and will not coordinate or be in communication with the Advisor including with respect to any decision by the Advisor to delay or proceed with trading.
Similar to a Nasdaq-listed firm-commitment underwritten initial public offering, in connection with the listing of our shares of common stock, buyers and sellers who have subscribed will have access to Nasdaq’s Order Imbalance Indicator, or the Net Order Imbalance Indicator, a widely available, subscription-based data feed, prior to submitting buy or sell orders. Nasdaq’s electronic trading platform simulates auctions every second to calculate a Current Reference Price, the number of shares of common stock that can be paired off the Current Reference Price, the number of shares of common stock that would remain unexecuted at the Current Reference Price and whether a buy-side or sell-side imbalance exists, or whether there is no imbalance, to disseminate that information continuously to buyers and sellers via the Net Order Imbalance Indicator data feed.
However, because this is not an initial public offering being conducted on a firm-commitment underwritten basis, there will be no traditional book building process (that is, an organized process pursuant to which buy and sell interest is coordinated in advance to some prescribed level – the “book”). Moreover, prior to the opening trade, there will not be a price at which underwriters initially sold shares of common stock to the public, as there would be in a firm-commitment underwritten initial public offering. The lack of an initial public offering price could impact the range of buy and sell orders collected by Nasdaq from various broker-dealers. Consequently, the public price of our shares of common stock may be more volatile than in an initial public offering underwritten on a firm-commitment basis and could, upon being listed on Nasdaq, decline significantly and rapidly.
In addition, to list on Nasdaq, we are also required to have at least four registered and active market makers. We expect that the Advisor will act as a registered and active market maker and will engage other market makers.
In addition to sales made pursuant to this prospectus, the shares of common stock covered by this prospectus may be sold by the Registered Stockholders in private transactions exempt from the registration requirements of the Securities Act. Under the securities laws of some states, shares of common stock may be sold in such states only through registered or licensed brokers or dealers.
| 127 |
A Registered Stockholder may from time to time transfer, distribute (including distributions in kind by Registered Stockholders that are investment funds), pledge, assign, or grant a security interest in some or all the shares of common stock owned by it and, if it defaults in the performance of its secured obligations, the transferees, distributees, pledgees, assignees, or secured parties may offer and sell the shares of common stock from time to time under this prospectus, or under an amendment to this prospectus under applicable provisions of the Securities Act amending the list of the Registered Stockholders to include the transferee, distributee, pledgee, assignee, or other successors in interest as Registered Stockholders under this prospectus. The Registered Stockholders also may transfer the shares in other circumstances, in which case the transferees, distributes, pledgees, or other successors in interest will be the registered beneficial owners for purposes of this prospectus.
A Registered Stockholder that is an entity may elect to make an in-kind distribution of common stock to its members, partners, or stockholders pursuant to the registration statement of which this prospectus forms a part by delivering a prospectus.
If any of the Registered Stockholders utilize a broker-dealer in the sale of the shares of common stock being offered by this prospectus, such broker-dealer may receive commissions in the form of discounts, concessions or commissions from such Registered Stockholder or commissions from purchasers of the shares of common stock for whom they may act as agent or to whom they may sell as principal.
We have engaged the Advisor, Maxim Group LLC, as our financial advisor to advise and assist us with respect to certain matters relating to the Direct Listing. The services expected to be performed by the Advisor will include providing advice and assistance with respect to defining objectives, analyzing, structuring and planning the Direct Listing and developing and assisting with our investor communication strategy in relation to the Direct Listing. In connection with its engagement as our financial advisor, the Advisor will be entitled to a fee of $200,000 upon the successful consummation of the Direct Listing. The Advisor will also be entitled to an expense reimbursement for all reasonable, documented expenses incurred by the Advisor in connection with its engagement, provided that (i) such expenses, other than legal fees, may not exceed $5,000 without our prior authorization and (ii) such expenses that constitute legal fees may not exceed $10,000 without our prior authorization.
In addition, pursuant to our agreement with the Advisor, for a period of nine months from the date of the consummation of the Direct Listing, if we propose to (i) effect a public offering of our securities on a major U.S. exchange, (ii) effect a private placement of our securities, (iii) enter into certain financing transactions with third parties introduced to us by the Advisor or (iv) propose to enter into certain other transactions with third parties introduced to us by the Advisor, including, without limitation, a merger, acquisition or sale of stock or assets, or other similar transaction, we are obligated to offer to retain the Advisor as our lead underwriter and book running manager, our lead placement or sales agent, or our lead advisor (lead, but not exclusive, advisor with at least 70% economics to lead and up to 30% to others), as applicable, in connection with such financing or transaction, upon such reasonable and customary terms as the Advisor and we may mutually agree, with such terms to be set forth in a separate engagement letter or other agreement between the Advisor and us.
The Advisor will not be engaged to otherwise facilitate or coordinate price discovery activities or the solicitation and/or sales of shares of our common stock in consultation with us, and will not be permitted to, and will not be instructed by us to, plan or actively participate in any investor education activities, except as described herein.
Prior to the financial advisory services provided by the Advisor to us in connection with the listing of our securities, neither the Advisor nor any affiliates of the Advisor have provided services of any kind to us.
| 128 |
The validity of the shares of common stock offered hereby will be passed upon for us by Norton Rose Fulbright US LLP, Houston, Texas.
The audited financial statements of FibroBiologics, Inc. as of and for the years ended December 31, 2022 and 2021 included in this registration statement have been audited by Withum Smith+Brown, PC, an independent registered public accounting firm, as stated in their report appearing herein. Such audited financial statements have been so included in reliance upon the report of such firm given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act, with respect to the shares of common stock being offered by this prospectus. This prospectus, which constitutes part of the registration statement, does not contain all of the information set forth in the registration statement or the exhibits and schedules filed therewith. Statements contained in this prospectus regarding the contents of any contract or any other document that is filed as an exhibit to the registration statement are not necessarily complete, and each such statement is qualified in all respects by reference to the full text of such contract or other document filed as an exhibit to the registration statement.
Immediately upon the effectiveness of the registration statement of which this prospectus forms a part, we will become subject to the information and reporting requirements of the Exchange Act and, in accordance with such law, will file annual, quarterly and current reports, proxy statements and other information with the SEC. The SEC maintains a website that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC. You may obtain documents that we file with the SEC at www.sec.gov. Our website address is www.fibrobiologics.com. We do not incorporate the information on or accessible through our website into this prospectus, and you should not consider any information on, or that can be accessed through, our website as part of this prospectus. Our website address is included in this prospectus as an inactive textual reference only.
| 129 |
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of
FibroBiologics, Inc.:
Opinion on the Financial Statements
We have audited the accompanying balance sheets of FibroBiologics, Inc. (the “Company”) as of December 31, 2022 and 2021, and the related statements of operations, changes in stockholders’ deficit, and cash flows for each of the two years in the period ended December 31, 2022, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2022 and 2021, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB and in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ WithumSmith+Brown PC
We have served as the Company’s auditor since 2022.
East Brunswick, New Jersey
April 28, 2023, except for the effects of the reverse stock split described in Note 13, as to which the date is November 7, 2023
| F-2 |
FibroBiologics, Inc.
(in thousands, except shares and per share data)
| December 31, | ||||||||
| 2022 | 2021 | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 2,266 | $ | 407 | ||||
| Prepaid expenses | 29 | 37 | ||||||
| Parent company receivable | 300 | — | ||||||
| Other current assets | 30 | 24 | ||||||
| Total current assets | 2,625 | 468 | ||||||
| Operating lease right-of-use asset, net | 2,199 | — | ||||||
| Total assets | $ | 4,824 | $ | 468 | ||||
| Liabilities and stockholders’ deficit | ||||||||
| Current liabilities | ||||||||
| Accounts payable and accrued expenses | $ | 758 | $ | 233 | ||||
| Parent company payable | — | 225 | ||||||
| Operating lease liability, short-term | 326 | — | ||||||
| Derivative liability | 538 | — | ||||||
| Convertible notes payable, net of debt discount | 5,451 | 1,300 | ||||||
| Total current liabilities | 7,073 | 1,758 | ||||||
| Operating lease liability, long-term | 1,747 | — | ||||||
| Total liabilities | 8,820 | 1,758 | ||||||
| Stockholders’ deficit | ||||||||
| Net Parent investment | 1,461 | 1,461 | ||||||
| Preferred Stock, $0.00001 par; 12,500,000 total shares authorized; 8,750,000 Series “A” Preferred shares authorized, issued and outstanding as of December 31, 2022 and 2021 | — | — | ||||||
| Preferred Stock, $0.00001 par; 12,500,000 total shares authorized; 2,500,000 Series “B” Preferred shares authorized; 381,658 shares issued and outstanding as of December 31, 2022; no shares issued and outstanding as of December 31, 2021 | — | — | ||||||
| Non-voting Common Stock, $0.00001 par; 62,500,000 shares authorized; 28,230,842 shares issued and outstanding as of December 31, 2022; no shares issued and outstanding as of December 31, 2021 | 1 | — | ||||||
| Additional paid-in capital | 2,414 | — | ||||||
| Accumulated deficit | (7,872 | ) | (2,751 | ) | ||||
| Total stockholders’ deficit | (3,996 | ) | (1,290 | ) | ||||
| Total liabilities and stockholders’ deficit | $ | 4,824 | $ | 468 | ||||
The accompanying notes are an integral part of these financial statements.
| F-3 |
FibroBiologics, Inc.
(in thousands, except shares and per share data)
| For the Years Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | 1,147 | $ | 521 | ||||
| General, administrative and other | 3,320 | 1,057 | ||||||
| Total operating expenses | 4,467 | 1,578 | ||||||
| Loss from operations | (4,467 | ) | (1,578 | ) | ||||
| Interest expense | (654 | ) | (4 | ) | ||||
| Net loss | $ | (5,121 | ) | $ | (1,582 | ) | ||
| Net loss per share, basic and diluted | $ | (.18 | ) | $ | N/A | |||
| Weighted-average shares outstanding, basic and diluted | 28,230,842 | N/A | ||||||
The accompanying notes are an integral part of these financial statements.
| F-4 |
FibroBiologics, Inc.
Statements of Changes in Stockholders’ Deficit
For the years ended December 31, 2022 and 2021
(in thousands, except shares)
| Net Parent |
Series “A” Preferred Stock |
Series “B” Preferred Stock |
Non-voting Common Stock |
Additional Paid-in | Accumulated |
Total Stockholders |
||||||||||||||||||||||||||||||||||
| Investment | Shares | Amount | Shares | Amount | Shares | Amount | Capital | Deficit | Deficit | |||||||||||||||||||||||||||||||
| Balance – December 31, 2020 | $ | 1,169 | — | $ | — | — | $ | — | — | $ | — | $ | — | $ | (1,169 | ) | $ | — | ||||||||||||||||||||||
| Issuance of capital shares upon Company formation | — | 8,750,000 | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||
| Capital contributions | 292 | — | — | — | — | — | — | — | — | 292 | ||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | (1,582 | ) | (1,582 | ) | ||||||||||||||||||||||||||||
| Balance – December 31, 2021 | 1,461 | 8,750,000 | — | — | — | — | — | — | (2,751 | ) | (1,290 | ) | ||||||||||||||||||||||||||||
| Issuance of Non-Voting Common Stock to Parent company members | — | — | — | — | — | 28,179,592 | 1 | (1 | ) | — | — | |||||||||||||||||||||||||||||
| Issuance of Series “B” Preferred shares | — | — | — | 381,658 | — | — | — | 2,150 | — | 2,150 | ||||||||||||||||||||||||||||||
| Stock-based compensation expense | — | — | — | — | — | 51,250 | — | 265 | — | 265 | ||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | (5,121 | ) | (5,121 | ) | ||||||||||||||||||||||||||||
| Balance – December 31, 2022 | $ | 1,461 | 8,750,000 | $ | — | 381,658 | $ | — | 28,230,842 | $ | 1 | $ | 2,414 | $ | (7,872 | ) | $ | (3,996 | ) | |||||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
| F-5 |
FibroBiologics, Inc.
(in thousands)
| For the Years Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (5,121 | ) | $ | (1,582 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation expense |
265 | — | ||||||
| Amortization of convertible notes debt discount |
389 | — | ||||||
| Amortization of operating lease right-of-use asset | 94 | — | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Change in prepaid expenses | 8 | (37 | ) | |||||
| Change in accounts payable and accrued expenses | 525 | 233 | ||||||
| Change in other current assets | (6 | ) | (24 | ) | ||||
| Change in operating lease liability | (220 | ) | — | |||||
| Net cash used in operating activities | (4,066 | ) | (1,410 | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from borrowing from Parent | — | 975 | ||||||
| Repayment to Parent | (225 | ) | (750 | ) | ||||
| Loan to Parent | (360 | ) | — | |||||
| Repayment from Parent | 60 | |||||||
| Proceeds from net Parent investment | — | 292 | ||||||
| Proceeds from issuance of convertible notes | 4,300 | 1,300 | ||||||
| Proceeds from issuance of Series “B” Preferred Stock | 2,150 | — | ||||||
| Net cash provided by financing activities | 5,925 | 1,817 | ||||||
| Net increase in cash and cash equivalents | 1,859 | 407 | ||||||
| Cash and cash equivalents, beginning of year | 407 | — | ||||||
| Cash and cash equivalents, end of year | $ | 2,266 | $ | 407 | ||||
| Supplemental disclosure of cash flow information: | ||||||||
| Cash paid for income taxes | $ | — | $ | — | ||||
| Cash paid for interest | $ | — | $ | — | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Addition to derivative liability for debt issuance discount | $ | 538 | $ | — | ||||
| Obtaining operating lease right-of-use asset and liability | $ | 2,293 | $ | — | ||||
The accompanying notes are an integral part of these financial statements.
| F-6 |
FibroBiologics, Inc.
Notes to the Financial Statements
December 31, 2022
| 1. | Organization, Description of Business, and Liquidity |
Organization and Business
FibroBiologics, Inc. (the “Company”, “FibroBiologics”) was originally formed as an LLC under the laws of the State of Texas on April 8, 2021 (“Inception”) and then converted to a Delaware corporation on December 14, 2021. FibroBiologics is an early stage, cell therapy company headquartered in Houston, Texas, developing innovative treatments for chronic diseases using fibroblast cells. The Company’s primary focus is the initiation and progression of preclinical studies and clinical-stage FDA trials related to fibroblast treatments for Degenerative Disc Disease, Multiple Sclerosis, Cancer, Wound Healing and other diseases. Prior to Inception, preclinical research and development related to these disease pathways took place under our parent company, SpinalCyte, LLC (the “Parent”, “FibroGenesis”).
Going Concern and Management’s Plan
The Company has incurred operating losses since Inception and expects such losses to continue in the future as it builds infrastructure, develops intellectual property and conducts research and development activities. The Company has primarily relied on a combination of angel investors and private debt placements to funds its operations. As of December 31, 2022, the Company had an accumulated deficit of $7,872 thousand and cash and cash equivalents of $2,266 thousand. A transition to profitability will depend on the successful development, approval and commercialization of product candidates and on the achievement of sufficient revenues to support the Company’s cost structure. The Company currently does not generate revenues and may never achieve profitability. Unless and until such time that revenue and net income are generated, the Company will need to continue to raise additional capital. As further described in Note 7, management has entered into a share purchase agreement as of November 12, 2021. In the event of a direct listing or an initial public offering on a nationally recognized U.S. stock exchange, this agreement will provide the Company with access to additional liquidity. As further described in Note 12, during the first three months of 2023 the Company raised $5 million through a crowdfunding offering and more than $10 million through a private placement offering. As a result, the Company believes it has adequate capital to fund its current operating plan for at least the next 12 months from the date of issuance of these Financial Statements.
Segments
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker in making decisions regarding resource allocation and assessing performance. The chief executive officer, who is the chief operating decision maker, reviews financial information on an aggregate basis for purposes of allocating resources and evaluating financial performance. The Company operates and manages its business as a single operating segment and therefore one reportable segment.
| 2. | Summary of Significant Accounting Policies |
Basis of Presentation
During the initial period during 2021 prior to its formation on April 8, 2021, the Company operated as a line of business of FibroGenesis rather than as a separate stand-alone entity. Consequently, stand-alone financial statements were not historically prepared for FibroBiologics. These Financial Statements have been prepared in connection with the formation and planned public listing of FibroBiologics and, prior to the Company’s formation on April 8, 2021, were derived from the historical accounting records of the Parent. All expenses, assets, and liabilities directly associated with the business activity of the Company as well as certain allocations from the Parent are included in the Financial Statements. Such allocations include the Company’s portion of general and administrative expenses and research and development expenses originally incurred by the Parent prior to the Company’s formation on April 8, 2021, for the disease pathways now pursued by FibroBiologics.
The expense allocations were determined by management and derived from the number of patents transferred to the Company through the patent transfer and assignment agreement between FibroBiologics and FibroGenesis. Patents were determined to be the most reasonable basis for allocation because patent development is the main driver of business activity for each entity during the preclinical phase, and they are the strongest proxy for expenses incurred by the Parent on behalf of the Company. Management believes the assumptions underlying the Financial Statements, including the assumptions regarding the allocation of expenses from the Parent, are reasonable. However, amounts recognized by the Company are not necessarily representative of the amounts that would have been reflected in the Financial Statements had the Company operated independently of the Parent as a standalone entity during the periods presented.
The accompanying Financial Statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”).
| F-7 |
Net Parent Investment
Because the Financial Statements are derived from the historical records of the Parent, the net Parent investment is presented within stockholders’ deficit on the Balance Sheets. As a subsidiary of the Parent, the Company was dependent upon the Parent for all of its working capital and financing requirements prior to entering into the Convertible Note agreements. Financial transactions that relate to FibroBiologics but occurred at the Parent level are accounted for through the net Parent investment account. Accordingly, none of the Parent’s cash, cash equivalents, or debt has been assigned to the Company in the financial statements. Net Parent investment represents the Parent’s interest in the recorded net assets of the Company.
Use of Estimates
The preparation of the Financial Statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the Financial Statements and the reported amounts of expenses during the reporting periods. These estimates are based on information available as of the date of the Financial Statements; therefore, actual results could differ from those estimates and assumptions.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash and cash equivalents. The Company has significant cash balances at financial institutions, which, throughout the year, regularly exceed the federally insured limit of $250,000. Any loss incurred or a lack of access to such funds could have a significant adverse impact on the Company’s financial condition, results of operations and cash flows.
Risks and Uncertainties
The Company is subject to certain risks and uncertainties, including, but not limited to changes in any of the following areas that the Company believes could have a material adverse effect on the future financial position or results of operations: the timing of, and the Company’s ability to advance its current and future product candidates into and through clinical development; costs and timelines associated with the manufacture of clinical supplies of the Company’s product candidates; regulatory approval and market acceptance of its product candidates; performance of third-party contract research organizations (“CROs”) and contract manufacturing organizations (“CMOs”); competition from pharmaceutical companies with greater financial resources or expertise; protection of the intellectual property, litigation or claims against the Company based on intellectual property, or other factors; the need to obtain additional funding; and its ability to attract and retain employees necessary to support its growth. Disruption from CROs’, CMOs’ or suppliers’ operations would likely have a negative impact on the Company’s business, financial position and results of operations.
Cash and Cash Equivalents
Cash and cash equivalents consist of unrestricted cash balances and short-term, liquid investments with an original maturity date of three months or less at the time of purchase.
Fair Value Measurements
Accounting Standards Codification (“ASC”) Topic 820, Fair Value Measurement (“ASC 820”), establishes a fair value hierarchy for instruments measured at fair value that distinguishes between assumptions based on market data (observable inputs) and the Company’s own assumptions (unobservable inputs). Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the inputs that market participants would use in pricing the assets or liability and are developed based on the best information available in the circumstances. ASC 820 identifies fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. As a basis for considering market participant assumptions in fair value measurements, ASC 820 establishes a three-tiered value hierarchy that distinguishes between the following:
Level 1 - Quoted market prices in active markets for identical assets or liabilities.
Level 2 - Inputs other than Level 1 inputs that are either directly or indirectly observable, such as quoted market prices, interest rates and yield curves.
Level 3 - Unobservable inputs for the asset or liability (i.e., supported by little or no market activity). Level 3 inputs include management’s own assumptions about the assumptions that market participants would use in pricing the asset or liability (including assumptions about risk).
| F-8 |
Research and Development
Research and development costs are charged to expense as incurred. Research and development costs consist of costs incurred in performing research and development activities, including salaries and bonuses, scientist recruiting costs, employee benefits, facilities costs, laboratory supplies, manufacturing expenses, preclinical expenses, research materials, and consulting and other contracted services. Costs for certain research and development activities are recognized based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the Financial Statements as prepaid or accrued research and development.
Patent Costs
As the Company continues to incur costs to obtain market approval of patented technology, patent costs are expensed as incurred. Costs include fees to renew or extend the term of recognized intangible assets, patent defense costs, and patent application costs. Management will continue to expense such costs until market approval is obtained through regulatory approval by the appropriate governing body.
Income Taxes
On December 8, 2021, the Company converted from a partnership LLC to a C-Corp. Subsequent to this date, the Company began accounting for income taxes under the asset and liability method. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Valuation allowances are established when necessary to reduce deferred tax assets to an amount that is more likely than not to be realized.
Under the provisions of ASC 740-10, Income Taxes, the Company evaluates uncertain tax positions by reviewing against applicable tax law all positions taken by the Company with respect to tax years for which the statute of limitations is still open. ASC 740-10 provides that a tax benefit from an uncertain tax position may be recognized when it is more likely than not that the position will be sustained upon examination, including resolutions of any related appeals or litigation processes, based on the technical merits. The Company recognizes interest and penalties related to the liability for unrecognized tax benefits, if any, as a component of the income tax expense line in the accompanying Statements of Operations.
Recently Adopted Accounting Pronouncements
In August 2020, the FASB issued ASU No. 2020-06, Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity (ASU 2020-06), which simplifies the accounting for convertible instruments by reducing the number of accounting models available for convertible debt instruments. This guidance also eliminates the treasury stock method to calculate diluted earnings per share for convertible instruments and requires the use of the if-converted method. The Company has early adopted this standard as of January 1, 2021, which is currently reflected in its Financial Statements.
In December 2019, the FASB issued ASU No. 2019-12, Income Taxes (Topic 740). The amendments in ASU No. 2019-12 simplify the accounting for income taxes by removing certain exceptions to the general principles in Topic 740. The amendments also improve consistent application of and simplify U.S. GAAP or other areas of Topic 740 by clarifying and amending existing guidance. The new standard was effective for the Company on January 1, 2022, and for interim periods beginning on January 1, 2023. The Company has adopted this standard which is currently reflected in its Financial Statements.
| 3. | Net Loss Per Share Attributable to Common Stockholders |
The following table summarizes the computation of basic and diluted net loss per share attributable to common stockholders of the Company:
| Year Ended December 31, | ||||||||
| (in thousands, except share and per share amounts) | 2022 | 2021 | ||||||
| Numerator: | ||||||||
| Net loss attributable to common stockholders: | $ | (5,121 | ) | $ | (1,582 | ) | ||
| Denominator: | ||||||||
| Weighted-average number of common shares outstanding, basic and diluted | 28,230,842 | N/A | ||||||
| Net loss per common share attributable to common stockholders, basic and diluted (1) | $ | (0.18 | ) | $ | N/A | |||
(1) The Company had no shares of its common stock issued and outstanding during the year ended December 31, 2021.
| F-9 |
As further described in Note 6, the Company issued 28,230,842 shares of non-voting common stock on August 18, 2022, and 381,658 shares of Series “B” Preferred Stock in December 2022. The weighted average number of shares outstanding for the year ended December 31, 2022, is based upon the non-voting common stock shares issued on August 18, 2022.
The Company had $5,600 thousand of convertible notes outstanding as of December 31, 2022, which may be converted into common stock in the event that the Company issues and sells shares of its capital stock in excess of $10,000 thousand as further described in Note 5. As of December 31, 2022, the estimated number of shares of common stock that would be issued upon conversion is 801,145 shares. For the years ended December 31, 2022 and 2021, the Company reported net losses and, accordingly, potential common shares were not included since such inclusion would have been anti-dilutive. As a result, the Company’s basic and diluted net losses per share are the same because it generated a net loss in all periods presented.
| 4. | Fair Value of Financial Instruments |
As of December 31, 2022, the Company measures its derivative liability related to the conversion option feature in the 2022 Notes, as described in Note 5, at fair value. This derivative liability is classified within Level 3 of the value hierarchy because the liability is based upon a valuation model that uses inputs and assumptions including potential outcomes, interest rates, probabilities, and timing. As of December 31, 2021, the Company did not have any financial instruments measured at fair value on a recurring basis.
The carrying amounts of cash and cash equivalents, prepaid expenses, other current assets, accounts payable, accrued expenses, convertible notes payable, and Parent company payable and receivable approximate their fair values due to their short-term maturities.
There were no transfers in or out of Level 1, Level 2 or Level 3 assets and liabilities for the years ended December 31, 2022 and 2021.
| 5. | Convertible Notes Payable |
The Company entered into multiple convertible promissory note agreements in December 2021 (collectively the “2021 Notes”). Under the 2021 Notes, the Company received $1,300 thousand, which accrues simple interest at a rate of 6.0% per annum and matures in the event of an initial public offering of the Company. Upon maturity of the 2021 Notes, the holders may elect to receive cash payment in full for the outstanding principal and interest or elect a one-year extension at the discretion of the holders of the 2021 Notes.
In the event that the Company issues and sells shares of its capital stock in excess of $10,000 thousand, the outstanding balance of the 2021 Notes and accrued interest may be converted into a fixed number of shares of common stock, subject only to typical anti-dilution provisions for any recapitalization that may occur.
Based on the terms of the 2021 Notes, the Company evaluated the conversion option feature in accordance with ASC 815 Derivatives and Hedging. It provides three criteria that, if met, require companies to bifurcate conversion options from their host instruments and account for them as freestanding derivative financial instruments. These three criteria include circumstances in which (a) the economic characteristics and risks of the embedded derivative instrument are not clearly and closely related to the economic characteristics and risks of the host contract, (b) the hybrid instrument that embodies both the embedded derivative instrument and the host contract is not re-measured at fair value under otherwise applicable generally accepted accounting principles with changes in fair value reported in earnings as they occur and (c) a separate instrument with the same terms as the embedded derivative instrument would be considered a derivative instrument.
At the inception of the 2021 Notes, and at December 31, 2021 and 2022, the Company determined that an embedded derivative for the conversion feature did not meet the criteria because it met the “indexed to the entity’s own stock” exception and therefore was not required to be bifurcated from the host instrument.
The Company issued additional convertible promissory notes between January and April 2022 with a total principal amount of $4,300 thousand and a one-year maturity (collectively the “2022 Notes”). The 2022 Notes may be converted at the lesser of a) a 15% discount to the offering price of the Company’s common stock in the event of an initial public offering of the Company or b) the quotient of $200,000 thousand divided by total equity interests prior to the dilution from the offering. The conversion option feature in the 2022 Notes was evaluated in accordance with ASC 815, and a derivative liability for the $538 thousand estimated fair value of the conversion option was recorded at the time the notes were issued and as of December 31, 2022. An offsetting discount on the issuance of the notes was recorded and is being amortized to interest expense over the expected life of the 2022 Notes.
The interest expense, excluding amortization of the discount recorded on the 2022 Notes, on the 2021 and 2022 Notes for the years ended December 31, 2022 and 2021, was $265 thousand and $4 thousand, respectively, which was outstanding and included within accounts payable and accrued expenses.
| F-10 |
The convertible debt balances consisted of the following at December 31, 2022 and 2021:
| December 31, | ||||||||
| (in thousands) | 2022 | 2021 | ||||||
| Convertible notes principal | $ | 5,600 | $ | 1,300 | ||||
| Convertible notes discount | (149 | ) | — | |||||
| Convertible notes payable, net of discount | $ | 5,451 | $ | 1,300 | ||||
| 6. | Stockholders’ Deficit / Net Parent Investment |
Authorized Capital - As of December 31, 2022 and 2021, the Company authorized 12,500,000 preferred stock shares, and has issued 8,750,000 Series “A” Preferred Stock shares to FibroGenesis, which were tendered pursuant to the formation of the Company in exchange for the contribution of certain in-process research and development and patent assets through Patent Assignment and Intellectual Property Cross-License Agreements. The Series “A” Preferred Stock shares have the right to vote and rank prior to non-voting common stock and common stock with respect to payment of dividends and distributions and upon liquidation, dissolution, winding-up or otherwise. In addition, the Series “A” Preferred Stock has a liquidation preference equal to $35,000 thousand to be allocated among the holders of the Series “A” Preferred Stock shares in the event of a liquidation, dissolution, or winding up of the Company, and each share of Series “A” Preferred Stock may be converted into one share of common stock at any time at the election of the holder of such shares of Series “A” Preferred Stock. Unless otherwise elected by the holder(s), a merger or consolidation in which the Company is not the majority surviving entity or the sale of all or substantially all of the assets of the corporation will be a deemed liquidation event. The Company has also authorized 62,500,000 shares of non-voting common stock, and has issued during the year ended December 31, 2022, a total of 28,230,842 shares. In August 2022, the Company issued 28,179,592 shares of non-voting common stock to its Parent, which in turn distributed the shares to its members. This issuance of non-voting common stock was accounted for as stock split and no proceeds were received by the Company. The Company also issued to its board of directors, a consultant, and an employee an additional 51,250 total shares in 2022 and recorded $168 thousand of expense for the issuance of these shares, which was based upon a third-party valuation of the shares at the time of issuance. None of the non-voting common stock shares were issued and outstanding as of December 31, 2021.
In December 2022, the Company amended its Certificate of Incorporation to authorize 2,500,000 shares of Series “B” Preferred Stock and issued 381,658 shares in exchange for $2,150 thousand. The Series “B” Preferred Stock has a liquidation preference after Series “A” Preferred Stock and prior to Common Stock and Non-Voting Common Stock. The Series “B” Preferred Stock has voting rights and will automatically convert into Common Stock upon closing of an IPO transaction, as defined in the Company’s Amended and Restated Certificate of Incorporation.
| 7. | Share Subscription Agreement |
On November 12, 2021, the Company entered into a Share Purchase Agreement with certain investors for the sale of up to $100,000 thousand of common stock (the “Aggregate Limit”). This agreement is contingent upon the Company achieving a public listing of its common stock. Major terms of the agreement include a commitment fee of 2% of the Aggregate Limit, which is due no later than one year after public listing even if no drawdowns are taken, and five-year warrants issued to the investors at the time of public listing to purchase common stock shares equal to 4% of the total equity interests of the Company at the lesser of a) the price per share at the time of the public listing or b) the quotient of $700,000 thousand divided by the total number of equity interests (fully diluted common shares). The Company may request a drawdown, or sale of common stock shares to the investors, over the five-year term of this agreement following the public listing unless terminated earlier. The amount of the drawdowns requested is limited by the trading volumes of the Company’s common stock shares over the 30-day period preceding the drawdown, and the price per share is equal to 90% of the average price per share over that same period. A 1% fee must be paid to the investors if the Company is sold in a private sale transaction rather than completing a public listing of its shares.
| F-11 |
| 8. | Income Taxes |
A reconciliation of the income tax benefit computed using the federal statutory income tax rate to the Company’s effective income tax rate is as follows:
| Year Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Federal statutory rate | (21.0 | )% | (21.0 | )% | ||||
| Permanent items | 0.8 | — | ||||||
| True up prior year NOL deferred tax asset | (6.1 | ) | — | |||||
| Other changes | 1.9 | — | ||||||
| Change in valuation allowance | 24.4 | 21.0 | % | |||||
| Total | 0.0 | % | 0.0 | % | ||||
The components of the Company’s net deferred tax assets are as follows:
| (in thousands of dollars) | December 31, 2022 | December 31, 2021 | ||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 896 | $ | — | ||||
| Lease liability | 435 | |||||||
| Capitalized research and development | 299 | |||||||
| Derivative liability | 31 | |||||||
| Accrued liabilities | 81 | 17 | ||||||
| Stock compensation | 17 | |||||||
| Deferred tax assets | 1,759 | 17 | ||||||
| Deferred tax liabilities: | ||||||||
| Lease right-of-use asset | (462 | ) | — | |||||
| Unamortized debt discount | (31 | ) | — | |||||
| Deferred tax liabilities | (493 | ) | — | |||||
| Less: valuation allowance | (1,266 | ) | (17 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
The Company was initially formed as an LLC and was converted to a Delaware corporation in December 2021. As a result of generating net operating losses during the years ended December 31, 2022 and 2021, the Company had no income tax expense for years ended December 31, 2022 and 2021. As of December 31, 2022, the Company had U.S. federal net operating loss (NOL) carryforwards of $4,265 thousand. The federal NOL carries forward indefinitely and may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest. This could limit the amount of tax attributes that can be utilized annually to offset future taxable income or tax liabilities. Subsequent ownership changes may further affect the limitation in future years.
Effective for tax years beginning after December 31, 2021, taxpayers are required to capitalize any expenses incurred that are considered incidental to research and experimentation (R&E) activities under IRC Section 174. While taxpayers historically had the option of deducting these expenses under IRC Section 174, the December 2017 Tax Cuts and Jobs Act mandates capitalization and amortization of R&E expenses for tax years beginning after December 31, 2021. Expenses incurred in connection with R&E activities in the US must be amortized over a 5-year period if incurred, and R&E expenses incurred outside the US must be amortized over a 15-year period. R&E activities are broader in scope than qualified research activities considered under IRC Section 41 (relating to the research tax credit). For the year ended December 31, 2022, the Company performed an analysis based on available guidance and determined that it will continue to be in a loss position even after the required capitalization and amortization of its R&E expenses. The Company will continue to monitor this issue for future developments, but it does not expect R&E capitalization and amortization to require it to pay cash taxes now or in the near future. We have included the impact of this provision, which results in a deferred tax asset of approximately $299 thousand as of December 31, 2022.
| F-12 |
Management has evaluated the positive and negative evidence bearing upon the realizability of the Company’s net deferred tax assets and has determined that it is more likely than not that the Company will not recognize the benefits of the net deferred tax assets. As a result, the Company has recorded a full valuation allowance at December 31, 2022 and 2021. The Company will continue to assess the realizability of its deferred tax assets going forward and will adjust the valuation allowance as needed.
As of December 31, 2022 and 2021, the Company had no uncertain tax positions. The Company recognizes both interest and penalties associated with unrecognized tax benefits as a component of income tax expense. The Company has not recorded any interest or penalties for unrecognized tax benefits since its inception.
| 9. | Leases, Commitments and Contingencies |
Effective January 1, 2020, the Company adopted ASU 2016-02, Leases (Topic 842) to account for the Company’s leases. The Company elected to apply the short-term lease practical expedient upon adoption. Due to the short-term nature of the leases, the Company elected an accounting policy to not record short-term leases on the Balance Sheets. ASC 842-20-25-2 allows a lessee to elect an accounting policy to not record short-term leases, defined as those with terms of 12 months or less, on the balance sheet. In accordance with GAAP, rent expense for financial statement purposes was recognized on a straight-line basis over the lease term based on the most recent contractual terms available.
As of December 31, 2021, the Company had entered into two short-term lease agreements for lab and office space. The Company opted on March 30, 2022, to extend the lease term for one of its leases for lab and office space, with a commencement date for the lease extension on May 1, 2022. The extended lease term was 126 months and was accounted for as an operating lease under the ASC 842 guidance for lease accounting. A right-of-use lease asset and tenant improvement allowance receivable with a combined total of $2,799 thousand and a lease liability of $2,799 thousand were recorded at the time of the extension. This lease was terminated as of July 31, 2022, and the remaining balances of the right-of-use asset, tenant improvement allowance receivable, and lease liability were written off.
The Company expanded the scope and extended for six months the term for the remaining lease for temporary lab and office space on July 1, 2022, and then further expanded the scope on August 1, 2022, and October 7, 2022. The monthly license fee increased to $15 thousand per month. This lease for temporary lab and office space will continue to be accounted for as a short-term lease.
In October 2022, the Company entered into a lease agreement for office space with a term of 62 months, which expires on November 30, 2027. This lease will be accounted for as an operating lease under the ASC 842 guidance for lease accounting. A right-of-use lease asset and lease liability of $2,293 thousand each were recorded at inception of the lease term using a discount rate of 7.5%.
Rent expense under operating leases for the years ended December 31, 2022 and 2021, was $392 thousand and $26 thousand, respectively. As of December 31, 2022, noncancelable lease payments were $2,484 thousand.
Maturities of operating lease liabilities as of December 31, 2022, were as follows:
| (in thousands of dollars) | ||||
| 2023 | $ | 466 | ||
| 2024 | 477 | |||
| 2025 | 488 | |||
| 2026 | 544 | |||
| 2027 | 509 | |||
| Thereafter | — | |||
| Total lease payments | 2,484 | |||
| Less: imputed interest | (411 | ) | ||
| Total lease liabilities | 2,073 | |||
| Less: current lease liabilities | (326 | ) | ||
| Total non-current lease liabilities | $ | 1,747 | ||
| 10. | Related Party Transactions |
As of December 31, 2021, the Company had an outstanding related party Parent company payable of $225 thousand. The debt was held by FibroGenesis, and was due April 1, 2022, with a six-month extension option available at the discretion of FibroBiologics. The Company repaid in April 2022 the remaining Parent company payable of $225 thousand. In July 2022, the Company loaned $300 thousand to the Parent on a one-year note bearing no interest. In October 2022, the Company loaned $60 thousand to the Parent on a one-year note bearing no interest and this note was repaid before December 31, 2022.
As described in Note 6, the Company acquired from FibroGenesis certain in-process research and development and patent assets through Patent Assignment and Intellectual Property Cross-License Agreements. The Patent Assignment Agreement transferred the right, title and interest in and to certain patents from FibroGenesis to the Company for further development. The Intellectual Property Cross-License Agreement grants to the Company exclusive rights to patents owned by FibroGenesis in a limited field of use, which includes the diagnosis, treatment, prevention and palliation of a) spinal diseases, disorders, or conditions, b) cancer, c) orthopedics diseases, disorders or conditions, and d) multiple sclerosis.
| F-13 |
| 11. | Share-based Compensation |
The Company adopted on August 10, 2022, and the shareholders approved on August 18, 2022, the 2022 Stock Plan (the “Plan”). The Plan provides for the grant of incentive stock options, nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, and other stock awards. The Plan, through the grant of stock awards, is intended to help the Company secure and retain the services of eligible award recipients, provide incentives for such persons to exert maximum efforts for the success of the Company and provide a means by which the eligible recipients may benefit from increases in value of the common stock. The Company issued in 2022 a total of 101,250 options with a strike price of $3.28 per share to employees, directors, and scientific advisory board members under this Plan. Generally, awards granted by the Company vest over three years and have an exercise price equal to the estimated fair value of the common stock as determined by the board of directors with consideration given to contemporaneous valuations of the Company’s common stock prepared by an independent third-party valuation firm.
As of December 31, 2022, there were 12,398,750 shares available for future issuance under the Plan.
Stock-based compensation expense is recognized in the Statements of Operations as follows:
| For the Years Ended December 31, | ||||||||
| (in thousands of dollars) | 2022 | 2021 | ||||||
| Research and development | $ | 115 | $ | — | ||||
| General and administrative | 150 | — | ||||||
| Total stock-based compensation expense | $ | 265 | $ | — | ||||
Stock-based compensation expense for the year ended December 31, 2022, includes $168 thousand of expense for non-voting common stock issued to the Board of Directors and consultants.
Unrecognized stock-based compensation costs related to unvested awards and the weighted-average period over which the costs are expected to be recognized as of December 31, 2022, are as follows:
| Stock Options | ||||
| Unrecognized stock-based compensation expense (in thousands) | $ | 169 | ||
| Expected weighted-average period compensation costs to be recognized (years) | 1.7 | |||
A summary of the Company’s stock option activity is as follows:
| Stock Options | Weighted-Average Exercise Price per Share | Weighted-Average Remaining Contractual Life (years) | Aggregate Intrinsic Value (in thousands) | |||||||||||||
| Outstanding as of December 31, 2021 | — | — | — | — | ||||||||||||
| Granted | 101,250 | $ | 3.28 | 10.0 | — | |||||||||||
| Exercised | — | — | — | — | ||||||||||||
| Forfeited/Canceled | — | — | — | — | ||||||||||||
| Outstanding as of December 31, 2022 | 101,250 | $ | 3.28 | 9.7 | — | |||||||||||
| Exercisable as of December 31, 2022 | 19,445 | $ | 3.28 | 9.7 | — | |||||||||||
The fair value of stock options granted to employees, directors, and consultants was estimated on the date of grant using the Black-Scholes option pricing model using the following assumptions:
| Assumptions: |
Year Ended December 31, 2022 |
|||
| Risk-free interest rate | 4.1 | % | ||
| Expected volatility | 100 | % | ||
| Expected term (years) | 5.4 to 6.4 | |||
| Expected dividend | 0 | % | ||
During the year ended December 31, 2022, the weighted-average grant date fair value of the options granted was $2.64 per share.
| F-14 |
| 12. | Subsequent Events |
The Company has evaluated subsequent events through April 28, 2023, the date the Financial Statements were available to be issued, and has determined that there were no other events, other than what is disclosed below, which occurred requiring disclosure in or adjustments to the Financial Statements.
In January 2023, the Company entered into an Agreement Regarding Right of First Negotiation (“ROFN Agreement”) with its Parent, FibroGenesis. In exchange for FibroGenesis’ consent to amend the Certificate of Incorporation to a) eliminate upon IPO, Direct Listing, or Sale of the Company the Series “A” Preferred Stock $35,000 thousand liquidation preference, b) make the Series “B” Preferred Stock liquidation preference equal to Series “A” Preferred Stock, and c) to provide that upon IPO, Direct Listing, or Sale of the Company Series “A” Preferred Stock will be canceled for no consideration, FibroBiologics will agree to pay to FibroGenesis 15% of the gross proceeds from any equity investments in FibroBiologics prior to an IPO, Direct Listing or Sale of the Company. In addition, FibroBiologics will receive a five-year right of first negotiation if FibroGenesis decides to license externally any of its technology. In January 2023, the Company amended its Certificate of Incorporation to reflect these changes and paid $323 thousand to FibroGenesis for 15% of the gross proceeds from equity issued by the Company in December 2022.
In January 2023, the Company launched a campaign to raise up to $5,000 thousand by selling Series “B” Preferred Stock through a Regulation CF offering, which was oversubscribed. This offering has closed with $4,990 thousand raised and the Company has received net proceeds of $4,230 thousand to date. The Company is in the process of receiving the remaining proceeds from this offering and anticipates issuing 867,913 shares when the final distributions are received and reconciled. Pursuant to the ROFN Agreement, the Company has paid $634 thousand (15%) of these proceeds to FibroGenesis in March 2023 and expects to pay an additional $114 thousand to FibroGenesis in April 2023 after the final distributions are received.
In January 2023, the Company extended for an additional six months its remaining lease for temporary lab and office space.
In February 2023, the Company converted the principal and interest on $3,700 thousand of principal value of the 2022 Notes into 799,603 shares of Series “B” Preferred Stock.
In March 2023, the Company sold an additional 1,680,084 shares of Series “B” Preferred Stock for $10,325 thousand in a private placement. Pursuant to the ROFN Agreement, the Company has paid $1,549 thousand (15%) of these proceeds to FibroGenesis.
In April 2023, the Company amended its Certificate of Incorporation to authorize 10,000,000 shares of Common Stock, increase the number of authorized Series “B” Preferred Stock shares up to 5,000,000 shares, and to authorize 5,000,000 shares of Series “B-1” Preferred Stock with liquidation preference equal to the Series “A” and “B” Preferred Stock. The Company also converted the principal and interest on $1,600 thousand of principal value of the 2021 Notes and $300 thousand of principal value on the 2022 Notes into 353,713 shares of Series “B” Preferred Stock and sold 8,388 shares of Series “B-1” Preferred Stock for $153 thousand in a private placement. Pursuant to the ROFN Agreement, the Company will pay $23 thousand (15%) of these proceeds to FibroGenesis in April 2023.
In April 2023, FibroGenesis repaid in full the $300 thousand Parent company receivable.
| 13. | Reverse Stock Split |
In October 2023, the Company filed an amended and restated certificate of incorporation with the State of Delaware to immediately effect a 1-for-4 reverse stock split. All share and per share amounts have been adjusted on a retroactive basis to reflect the effect of the reverse stock split.
| F-15 |
FibroBiologics, Inc.
Unaudited Condensed Financial Statements
and
Notes to the Unaudited Condensed Financial Statements
for the Nine Months Ended September 30, 2023 and 2022
| F-16 |
FibroBiologics, Inc.
(in thousands, except shares and per share data)
|
September 30, 2023 |
December 31, 2022 |
|||||||
| (Unaudited) | ||||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 10,766 | $ | 2,266 | ||||
| Prepaid expenses | 27 | 29 | ||||||
| Parent company receivable | — | 300 | ||||||
| Other current assets | 121 | 30 | ||||||
| Total current assets | 10,914 | 2,625 | ||||||
| Property and equipment, net | 477 | — | ||||||
| Operating lease right-of-use asset, net | 1,908 | 2,199 | ||||||
| Total assets | $ | 13,299 | $ | 4,824 | ||||
| Liabilities and stockholders’ equity/(deficit) | ||||||||
| Current liabilities | ||||||||
| Accounts payable and accrued expenses | $ | 820 | $ | 758 | ||||
| Payable to parent | 141 | — | ||||||
| Operating lease liability, short-term | 353 | 326 | ||||||
| Derivative liability | — | 538 | ||||||
| Convertible notes payable, net of debt discount | — | 5,451 | ||||||
| Total current liabilities | 1,314 |
7,073 | ||||||
| Operating lease liability, long-term | 1,447 | 1,747 | ||||||
| Total liabilities | 2,761 | 8,820 | ||||||
| Stockholders’ equity/(deficit) | ||||||||
| Net Parent investment | — | 1,461 | ||||||
| Preferred Stock, $0.00001 par; 20,000,000 total shares authorized; 8,750,000 Series A Preferred shares authorized, issued and outstanding as of September 30, 2023, and December 31, 2022 | — | — | ||||||
| Preferred Stock, $0.00001 par; 20,000,000 total shares authorized; 5,000,000 Series B Preferred shares authorized; 4,171,445 shares issued and outstanding as of September 30, 2023; 381,658 shares issued and outstanding as of December 31, 2022 | — | — | ||||||
Preferred Stock, $0.00001 par; 20,000,000 total shares authorized; 5,000,000 Series B-1 Preferred shares authorized; 74,922 shares issued and outstanding as of September 30, 2023; no shares issued and outstanding as of December 31, 2022 |
— | — | ||||||
| Preferred Stock, $0.00001 par; 20,000,000 total shares authorized; 2,500 Series C Preferred shares authorized; no shares issued and outstanding as of September 30, 2023 and December 31, 2022 | — |
— |
||||||
| Non-voting Common Stock, $0.00001 par; 30,000,000 shares authorized; 28,230,842 shares issued and outstanding as of September 30, 2023, and December 31, 2022 | 1 | 1 | ||||||
| Common Stock, $0.00001 par; 100,000,000 shares authorized; no shares issued and outstanding as of September 30, 2023, and December 31, 2022 | — | — | ||||||
| Additional paid-in capital | 25,177 | 2,414 | ||||||
| Accumulated deficit | (14,640 | ) | (7,872 | ) | ||||
| Total stockholders’ equity/(deficit) | 10,538 | (3,996 | ) | |||||
| Total liabilities and stockholders’ equity/(deficit) | $ | 13,299 | $ | 4,824 | ||||
The accompanying notes are an integral part of these Unaudited Condensed Financial Statements.
| F-17 |
FibroBiologics, Inc.
Condensed Statements of Operations
(unaudited, in thousands, except shares and per share data)
|
For the Nine Months Ended September 30, |
||||||||
| 2023 | 2022 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | 1,595 | $ | 802 | ||||
| General, administrative and other | 4,814 | 2,361 |
||||||
| Total operating expenses | 6,409 | 3,163 | ||||||
| Loss from operations | (6,409 | ) | (3,163 | ) | ||||
| Other income/(loss) | (213 | ) | — | |||||
| Interest expense | (146 | ) | (434 | ) | ||||
| Net loss | $ | (6,768 | ) | $ | (3,597 | ) | ||
| Deemed dividend | (2,573 | ) | — | |||||
| Net loss attributable to common stockholders | (9,341 | ) | $ | (3,597 | ) | |||
| Net loss per share, basic and diluted | $ | (.33 | ) | $ | (.13 | ) | ||
| Weighted-average shares outstanding, basic and diluted | 28,230,842 | 28,230,842 | ||||||
The accompanying notes are an integral part of these Unaudited Condensed Financial Statements.
| F-18 |
FibroBiologics, Inc.
Condensed Statements of Changes in Stockholders’ Equity/(Deficit)
For the Nine Months ended September 30, 2023 and 2022
(unaudited, in thousands, except shares)
| Net Parent | Series “A” Preferred Stock | Series “B” Preferred Stock | Series “B-1” Preferred Stock | Non-voting Common Stock | Additional Paid-in | Accumulated | Total Stockholders’ Equity/ |
|||||||||||||||||||||||||||||||||||||||||
| Investment | Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Capital | Deficit | (Deficit) | |||||||||||||||||||||||||||||||||||||
| Balance – December 31, 2022 | $ | 1,461 | 8,750,000 | $ | — | 381,658 | $ | — | — | $ | — | 28,230,842 | $ | 1 | $ | 2,414 | $ | (7,872 | ) | $ | (3,996 | ) | ||||||||||||||||||||||||||
| Sale of Series “B” Preferred Stock shares, net of direct costs | — | — | — | 2,570,394 | — | — | — | — | — | 14,945 | — | 14,945 | ||||||||||||||||||||||||||||||||||||
| Sale of Series “B-1” Preferred Stock shares, net of direct costs | — | — | — | — | — | 74,922 | — | — | — | 1,193 | — | 1,193 | ||||||||||||||||||||||||||||||||||||
| Issuance of Series “B” Preferred Stock shares for conversion of Notes and accrued interest | — | — | — | 1,219,393 | — | — | — | — | — | 6,404 | — | 6,404 | ||||||||||||||||||||||||||||||||||||
| Deemed dividend related to ROFN Agreement derivative liability | (1,461 | ) | — | — | — | — | — | — | — | — | (1,112 | ) | — | (2,573 | ) | |||||||||||||||||||||||||||||||||
| Stock-based compensation expense | — | — | — | — | — | — | — | — | — | 1,333 | — | 1,333 | ||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (6,768 | ) | (6,768 | ) | ||||||||||||||||||||||||||||||||||
| Balance (unaudited) – September 30, 2023 | $ | — | 8,750,000 | $ | — | 4,171,445 | $ | — | 74,922 | $ | — | 28,230,842 | $ | 1 | $ | 25,177 | $ | (14,640 | ) | $ | 10,538 | |||||||||||||||||||||||||||
| Net Parent | Series “A” Preferred Stock | Series “B” Preferred Stock | Series “B-1” Preferred Stock | Non-voting Common Stock | Additional Paid-in | Accumulated | Total Stockholders’ Equity/ |
|||||||||||||||||||||||||||||||||||||||||
| Investment | Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Capital | Deficit | (Deficit) | |||||||||||||||||||||||||||||||||||||
| Balance – December 31, 2021 | $ | 1,461 | 8,750,000 | $ | — | — | $ | — | — | $ | — | — | $ | — | $ | — | $ | (2,751 | ) | $ | (1,290 | ) | ||||||||||||||||||||||||||
| Issuance of Non-Voting Common Stock to Parent company members | — | — | — | — | — | — | — | 28,179,592 | 1 | (1 | ) | — | — | |||||||||||||||||||||||||||||||||||
| Stock-based compensation expense | — | — | — | — | — | — | — | 51,250 | — | 233 | — | 233 | ||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (3,597 | ) | (3,597 | ) | ||||||||||||||||||||||||||||||||||
|
Balance (unaudited) – September 30, 2022 |
$ | 1,461 | 8,750,000 | $ | — | — | $ | — | — | $ | — | 28,230,842 | $ | 1 | $ | 232 | $ | (6,348 | ) | $ | (4,654 | ) | ||||||||||||||||||||||||||
The accompanying notes are an integral part of these Unaudited Condensed Financial Statements.
| F-19 |
FibroBiologics, Inc.
Condensed Statements of Cash Flows
(unaudited, in thousands)
| For the Nine Months Ended September 30, | ||||||||
| 2023 | 2022 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (6,768 | ) | $ | (3,597 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation expense |
1,333 | 233 | ||||||
| Other loss on derivative liability | 72 | — | ||||||
| Amortization of convertible notes debt discount |
81 | 254 | ||||||
| Amortization of operating lease right-of-use asset | 291 | — | ||||||
| Depreciation expense | 16 | — | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Change in prepaid expenses | 2 | (21 |
) | |||||
| Change in other current assets | (91 | ) | (22 | ) | ||||
| Change in accounts payable and accrued expenses | 396 | 260 | ||||||
| Change in payable to Parent | 141 | — |
||||||
| Change in operating lease liability | (273 | ) | — | |||||
| Net cash used in operating activities | (4,800 | ) | (2,893 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchases of property and equipment | (493 | ) | — | |||||
| Net cash used in investing activities | (493 | ) | — | |||||
| Cash flows from financing activities | ||||||||
| Payment of loan to Parent | — | (225 | ) | |||||
| ROFN Agreement payments to Parent | (2,645 | ) | — | |||||
| Repayment and proceeds of note receivable from Parent | 300 | (300 |
) | |||||
| Proceeds from issuance of convertible notes | — | 4,300 | ||||||
| Proceeds from issuance of Series B Preferred Stock, net of direct costs | 14,945 | — | ||||||
| Proceeds from issuance of Series B-1 Preferred Stock, net of direct costs | 1,193 | — | ||||||
| Net cash provided by financing activities | 13,793 |
3,775 | ||||||
| Net increase in cash and cash equivalents | 8,500 |
882 | ||||||
| Cash and cash equivalents, beginning of period | 2,266 | 407 | ||||||
| Cash and cash equivalents, end of period | $ | 10,766 |
$ | 1,289 | ||||
| Supplemental disclosure of cash flow information: | ||||||||
| Cash paid for income taxes | $ | — | $ | — | ||||
| Cash paid for interest | $ | 1 | $ | — | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Addition to derivative liability for debt issuance discount | $ | — | $ | 538 | ||||
| Obtaining operating lease right-of-use asset and liability | $ | — | $ | 2,799 | ||||
| Issuance of Series B Preferred Stock for conversion of Notes and accrued interest | $ | 5,866 | $ | — | ||||
| Reclassification of derivative liability for conversion of Notes to Series B Preferred Stock | $ | 538 | $ | — | ||||
The accompanying notes are an integral part of these Unaudited Condensed Financial Statements.
| F-20 |
FibroBiologics, Inc.
Notes to the Unaudited Condensed Financial Statements
September 30, 2023
| 1. | Organization, Description of Business, and Liquidity |
Organization and Business
FibroBiologics, Inc. (the “Company” or “FibroBiologics”) was originally formed as a limited liability company (“LLC”) under the laws of the State of Texas on April 8, 2021 (“Inception”) and then converted to a Delaware corporation on December 14, 2021. FibroBiologics is an early stage, cell therapy company headquartered in Houston, Texas, developing innovative treatments for chronic diseases using fibroblast cells. The Company’s primary focus is the initiation and progression of preclinical studies and clinical-stage U.S. Food and Drug Administration trials related to fibroblast treatments for Degenerative Disc Disease, Multiple Sclerosis, Cancer, Wound Healing and other diseases. Prior to Inception, preclinical research and development related to these disease pathways took place under the parent company, SpinalCyte, LLC (the “Parent” or “FibroGenesis”).
Going Concern and Management’s Plan
The Company has incurred operating losses since Inception and expects such losses to continue in the future as it builds infrastructure, develops intellectual property and conducts research and development activities. The Company has primarily relied on a combination of angel investors and private debt placements to funds its operations. As of September 30, 2023, the Company had an accumulated deficit of $14,640 thousand and cash and cash equivalents of $10,766 thousand. A transition to profitability will depend on the successful development, approval and commercialization of product candidates and on the achievement of sufficient revenues to support the Company’s cost structure. The Company currently does not generate revenues and may never achieve profitability. Unless and until such time that revenue and net income are generated, the Company will need to continue to raise additional capital. As further described in Note 7, management has entered into a share purchase agreement as of November 12, 2021. In the event of a direct listing or an initial public offering on a nationally recognized U.S. stock exchange, this agreement will provide the Company with access to additional liquidity. As further described in Note 6, during the first nine months of 2023 the Company raised $4,620 thousand, net of fees, through a crowdfunding offering and $11,518 thousand through a private placement offering. As a result, the Company believes it has adequate capital to fund its current operating plan for at least the next 12 months from the date of issuance of these Unaudited Condensed Financial Statements.
Segments
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker in making decisions regarding resource allocation and assessing performance. The chief executive officer, who is the chief operating decision maker, reviews financial information on an aggregate basis for purposes of allocating resources and evaluating financial performance. The Company operates and manages its business as a single operating segment and therefore one reportable segment.
| 2. | Summary of Significant Accounting Policies |
Basis of Presentation
The accompanying Unaudited Condensed Financial Statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”). The accompanying condensed balance sheet as of September 30, 2023, condensed statements of operations for the nine months ended September 30, 2023 and 2022, condensed statements of stockholders’ equity/(deficit) for the nine months ended September 30, 2023 and 2022, and condensed statements of cash flows for the nine months ended September 30, 2023 and 2022, are unaudited. These Unaudited Condensed Financial Statements have been prepared in accordance with the rules and regulations of the United States Securities and Exchange Commission for interim financial information. Accordingly, they do not include all of the information and footnotes required by GAAP for complete financial statements. These Unaudited Condensed Financial Statements should be read in conjunction with the audited financial statements and the accompanying notes for the year ended December 31, 2022. The unaudited condensed financial statements have been prepared on the same basis as the annual consolidated financial statements and, in the opinion of management, reflect all adjustments (consisting of normal recurring adjustments) necessary to state fairly the Company’s financial position as of September 30, 2023, the results of operations for the nine months ended September 30, 2023 and 2022, the unaudited condensed statements of stockholders’ equity/(deficit) for the nine months ended September 30, 2023 and 2022 and the unaudited condensed statements of cash flows for the nine months ended September 30, 2023 and 2022. The December 31, 2022, condensed balance sheet included herein was derived from the audited financial statements, but it does not include all disclosures or notes required by GAAP for complete financial statements.
| F-21 |
The financial data and other information disclosed in these notes to the condensed financial statements related to the nine months ended September 30, 2023 and 2022, are unaudited. Interim results are not necessarily indicative of results for an entire year or for any future period.
During the period from January 1, 2021, to its formation on April 8, 2021, the Company operated as a line of business of FibroGenesis rather than as a separate stand-alone entity. Consequently, prior to the Company’s formation on April 8, 2021, the financial statements were derived from the historical accounting records of the Parent. All general and administrative expenses and research and development expenses directly associated with the business activity of the Company that were originally incurred by the Parent from January 1, 2021, through the Company’s formation on April 8, 2021, were allocated and included in the Company’s financial statements. The resulting net Parent investment is presented within stockholders’ equity/(deficit) and represents the Parent’s interest in the recorded net assets of the Company.
The accompanying Unaudited Condensed Financial Statements do not include any allocations from the Parent other than the net Parent investment included in the beginning stockholders’ equity/(deficit) and have been prepared in accordance with GAAP.
In October 2023, the Company amended and restated its certificate of incorporation with the State of Delaware to immediately effect a 1-for-4 shares reverse stock split. All share and per share amounts have been adjusted on a retroactive basis to reflect the effect of the reverse stock split.
Use of Estimates
The preparation of the Unaudited Condensed Financial Statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the Unaudited Condensed Financial Statements and the reported amounts of expenses during the reporting periods. These estimates are based on information available as of the date of the Unaudited Condensed Financial Statements; therefore, actual results could differ from those estimates and assumptions.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash and cash equivalents. The Company has significant cash balances at financial institutions, which, throughout the year, regularly exceed the federally insured limit of $250,000. Any loss incurred or a lack of access to such funds could have a significant adverse impact on the Company’s financial condition, results of operations and cash flows.
Risks and Uncertainties
The Company is subject to certain risks and uncertainties, including, but not limited to changes in any of the following areas that the Company believes could have a material adverse effect on the future financial position or results of operations: the timing of, and the Company’s ability to advance its current and future product candidates into and through clinical development; costs and timelines associated with the manufacture of clinical supplies of the Company’s product candidates; regulatory approval and market acceptance of its product candidates; performance of third-party contract research organizations (“CROs”) and contract manufacturing organizations (“CMOs”); competition from pharmaceutical companies with greater financial resources or expertise; protection of the intellectual property, litigation or claims against the Company based on intellectual property, or other factors; the need to obtain additional funding; and its ability to attract and retain employees necessary to support its growth. Disruption from the operations of CROs, CMOs or suppliers would likely have a negative impact on the Company’s business, financial position and results of operations.
Cash and Cash Equivalents
Cash and cash equivalents consist of unrestricted cash balances and short-term, liquid investments with an original maturity date of three months or less at the time of purchase.
Property and Equipment
Property and equipment include laboratory equipment that is recorded at cost and depreciated using the straight-line method over the estimated useful lives of five years. Depreciation expense of $16 thousand was recognized for the nine months ended September 30, 2023. No depreciation expense was recognized for the nine months ended September 30, 2022. Depreciation expense is classified in research and development expense in the accompanying Condensed Statements of Operations.
Property and equipment are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets might not be recoverable. Conditions that would necessitate an impairment assessment include a significant decline in the observable market value of an asset, a significant change in the extent or manner in which an asset is used, or a significant adverse change that would indicate that the carrying amount of an asset or group of assets is not recoverable. For long-lived assets to be held and used, the Company will recognize an impairment loss only if the carrying amount is not recoverable through its undiscounted cash flows and measure any impairment loss based on the difference between the carrying amount and estimated fair value. There were no such losses for the nine months ended September 30, 2023 and 2022.
| F-22 |
Fair Value Measurements
Accounting Standards Codification (“ASC”) 820, Fair Value Measurement, establishes a fair value hierarchy for instruments measured at fair value that distinguishes between assumptions based on market data (observable inputs) and the Company’s own assumptions (unobservable inputs). Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the inputs that market participants would use in pricing the assets or liability and are developed based on the best information available in the circumstances. ASC 820 identifies fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. As a basis for considering market participant assumptions in fair value measurements, ASC 820 establishes a three-tiered value hierarchy that distinguishes between the following:
Level 1 - Quoted market prices in active markets for identical assets or liabilities.
Level 2 - Inputs other than Level 1 inputs that are either directly or indirectly observable, such as quoted market prices, interest rates and yield curves.
Level 3 - Unobservable inputs for the asset or liability (i.e., supported by little or no market activity). Level 3 inputs include management’s own assumptions about the assumptions that market participants would use in pricing the asset or liability (including assumptions about risk).
Categorization within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement.
Research and Development
Research and development costs are charged to expense as incurred. Research and development costs consist of costs incurred in performing research and development activities, including salaries and bonuses, scientist recruiting costs, employee benefits, facilities costs, laboratory supplies, manufacturing expenses, preclinical expenses, research materials, and consulting and other contracted services. Costs for certain research and development activities are recognized based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the Unaudited Condensed Financial Statements as prepaid or accrued research and development.
Patent Costs
As the Company continues to incur costs to obtain market approval of patented technology, patent costs are expensed as incurred in general, administrative and other expense in the Unaudited Condensed Statements of Operations. Costs include fees to renew or extend the term of recognized intangible assets, patent defense costs, and patent application costs. Management will continue to expense such costs until market approval is obtained through regulatory approval by the appropriate governing body.
Income Taxes
On December 14, 2021, the Company converted from a partnership LLC to a C corporation. Subsequent to this date, the Company began accounting for income taxes under the asset and liability method. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Valuation allowances are established when necessary to reduce deferred tax assets to an amount that is more likely than not to be realized.
Under the provisions of ASC 740-10, Income Taxes, the Company evaluates uncertain tax positions by reviewing against applicable tax law all positions taken by the Company with respect to tax years for which the statute of limitations is still open. ASC 740-10 provides that a tax benefit from an uncertain tax position may be recognized when it is more likely than not that the position will be sustained upon examination, including resolutions of any related appeals or litigation processes, based on the technical merits. The Company recognizes interest and penalties related to the liability for unrecognized tax benefits, if any, as a component of the income tax expense line in the accompanying Condensed Statements of Operations.
| F-23 |
| 3. | Net Loss Per Share Attributable to Common Stockholders |
The following table summarizes the computation of basic and diluted net loss per share attributable to common stockholders of the Company:
|
Nine Months Ended September 30, |
||||||||
| (in thousands, except share and per share amounts) | 2023 | 2022 | ||||||
| Numerator: | ||||||||
| Net loss | $ | (6,768 | ) | $ | (3,597 | ) | ||
| Adjustment to numerator for earnings per share: | ||||||||
| Deemed dividend | (2,573 | ) | — | |||||
| Net loss attributable to common stockholders | $ | (9,341 | ) | $ | (3,597 | ) | ||
| Denominator: | ||||||||
| Weighted-average number of common shares outstanding, basic and diluted | 28,230,842 | 28,230,842 | ||||||
| Net loss per common share attributable to common stockholders, basic and diluted | $ | (0.33 | ) | $ | (0.13 | ) | ||
As further described in Note 6, the Company issued 28,230,842 shares of non-voting common stock on August 18, 2022. The weighted average number of shares outstanding for the nine months ended September 30, 2023 and 2022, is based upon the non-voting common stock shares issued on August 18, 2022. Because the issuance of nonvoting common stock shares was treated as a stock split for accounting purposes, these shares are treated as having been issued on January 1, 2022, and outstanding for the entire nine months ended September 30, 2023 and 2022.
As further described in Note 10, the Company agreed to pay to FibroGenesis 15% of the gross proceeds from any equity investments in FibroBiologics prior to an IPO, Direct Listing or Sale of the Company to eliminate upon the occurrence of such event the Series A Preferred Stock and its $35 million liquidation preference. This redemption of preferred stock created a derivative liability that exceeded the net Parent Investment of $1,461 thousand by $1,112 thousand, and is reflected here as a reduction of the amount available to common stockholders in the calculation of earnings per share.
The Company had $5,600 thousand of convertible notes outstanding as of September 30, 2022, which could have been converted into common stock in the event that the Company sold and issued shares of capital stock in excess of $10,000 thousand, and were all converted by September 30, 2023, into shares of Series B Preferred Stock as further described in Note 5. As of September 30, 2022, the estimated number of shares of common stock that would have been issued upon conversion was 790,459 shares. For the nine months ended September 30, 2023 and 2022, the Company reported net losses and, accordingly, potential common shares were not included since such inclusion would have been anti-dilutive. As a result, the Company’s basic and diluted net losses per share are the same because it generated a net loss in all periods presented.
| 4. | Fair Value of Financial Instruments |
As of December 31, 2022, the Company measured its derivative liability related to the conversion option feature in the 2022 Notes, as described in Note 5, at fair value. This derivative liability was classified within Level 3 of the value hierarchy because the liability was based upon a valuation model that used inputs and assumptions including potential outcomes, interest rates, probabilities, and timing. As of September 30, 2023, the 2022 Notes had been converted, which eliminated the derivative liability, and the Company did not have any material financial instruments measured at fair value on a recurring basis.
The carrying amounts of cash and cash equivalents, prepaid expenses, other current assets, accounts payable, accrued expenses, convertible notes payable, and Parent company payable and receivable approximate their fair values due to their short-term maturities.
There were no transfers in or out of Level 1, Level 2 or Level 3 assets and liabilities for the nine months ended September 30, 2023 and 2022.
| F-24 |
| 5. | Convertible Notes Payable |
The Company entered into multiple convertible promissory note agreements in December 2021 (collectively, the “2021 Notes”). Under the 2021 Notes, the Company received $1,300 thousand, which accrues simple interest at a rate of 6.0% per annum and matures in the event of an initial public offering of the Company. Upon maturity of the 2021 Notes, the holders may elect to receive cash payment in full for the outstanding principal and interest or elect a one-year extension at the discretion of the holders of the 2021 Notes.
In the event that the Company issues and sells shares of its capital stock in excess of $10,000 thousand, the outstanding balance of the 2021 Notes and accrued interest may be converted into a fixed number of shares of common stock, subject only to typical anti-dilution provisions for any recapitalization that may occur.
Based on the terms of the 2021 Notes, the Company evaluated the conversion option feature in accordance with ASC 815, Derivatives and Hedging. It provides three criteria that, if met, require companies to bifurcate conversion options from their host instruments and account for them as freestanding derivative financial instruments. These three criteria include circumstances in which (a) the economic characteristics and risks of the embedded derivative instrument are not clearly and closely related to the economic characteristics and risks of the host contract, (b) the hybrid instrument that embodies both the embedded derivative instrument and the host contract is not re-measured at fair value under otherwise applicable generally accepted accounting principles with changes in fair value reported in earnings as they occur and (c) a separate instrument with the same terms as the embedded derivative instrument would be considered a derivative instrument.
At the inception of the 2021 Notes, and at December 31, 2022, the Company determined that an embedded derivative for the conversion feature did not meet the criteria because it met the “indexed to the entity’s own stock” exception and therefore was not required to be bifurcated from the host instrument.
The Company issued additional convertible promissory notes between January and April 2022 with a total principal amount of $4,300 thousand and a one-year maturity (collectively, the “2022 Notes”). The 2022 Notes may be converted at the lesser of a) a 15% discount to the offering price of the Company’s common stock in the event of an initial public offering of the Company or b) the quotient of $200,000 thousand divided by total equity interests prior to the dilution from the offering. The conversion option feature in the 2022 Notes was evaluated in accordance with ASC 815, and a derivative liability for the $538 thousand estimated fair value of the conversion option was recorded at the time the notes were issued and as of December 31, 2022. An offsetting discount on the issuance of the notes was recorded and is being amortized to interest expense over the expected life of the 2022 Notes.
The interest expense, excluding amortization of the discount recorded on the 2022 Notes, on the 2021 and 2022 Notes for the nine months ended September 30, 2023 and 2022, was $65 thousand and $180 thousand, respectively. Accrued interest was outstanding and included within accounts payable and accrued expenses at December 31, 2022.
In February 2023, the Company converted the principal and interest on $3,700 thousand of principal value of the 2022 Notes into 799,603 shares of Series “B” Preferred Stock. In April 2023, the Company also converted the principal and interest on $1,300 thousand of principal value of the 2021 Notes and $300 thousand of principal value on the 2022 Notes into 353,713 shares of Series “B” Preferred Stock. In September 2023, the Company converted the principal and interest on $300 thousand of principal value of the 2022 Notes into 66,077 shares of Series “B” Preferred Stock. No interest was outstanding and included within accounts payable and accrued expenses at September 30, 2023.
The convertible debt balances consisted of the following at September 30, 2023, and December 31, 2022:
| (in thousands) | September 30, 2023 | December 31, 2022 | ||||||
| Convertible notes principal | $ | — | $ | 5,600 | ||||
| Convertible notes discount | — | (149 | ) | |||||
| Convertible notes payable, net of discount | $ | — | $ | 5,451 | ||||
| F-25 |
| 6. | Stockholders’ Equity/(Deficit) / Net Parent Investment |
Authorized Capital - As of September 30, 2023 and 2022, the Company authorized 20,000,000 and 12,500,000 preferred stock shares, respectively, and has issued 8,750,000 Series “A” Preferred Stock shares to FibroGenesis, which were tendered pursuant to the formation of the Company in exchange for the contribution of certain in-process research and development and patent assets through Patent Assignment and Intellectual Property Cross-License Agreements. The Series “A” Preferred Stock shares have the right to vote and rank prior to non-voting common stock and common stock with respect to payment of dividends and distributions and upon liquidation, dissolution, winding-up or otherwise. In addition, the Series “A” Preferred Stock had a liquidation preference equal to $35,000 thousand to be allocated among the holders of the Series “A” Preferred Stock shares in the event of a liquidation, dissolution, or winding-up of the Company, which was subsequently eliminated as part of the ROFN Agreement as further described below, and each share of Series “A” Preferred Stock may be converted into one share of common stock at any time at the election of the holder of such shares of Series “A” Preferred Stock. Unless otherwise elected by the holder(s), a merger or consolidation in which the Company is not the majority surviving entity or the sale of all or substantially all of the assets of the corporation will be a deemed liquidation event. The Company has also authorized 62,500,000 shares of non-voting common stock, and has issued during the year ended December 31, 2022, a total of 28,230,842 shares. In August 2022, the Company issued 28,179,592 shares of non-voting common stock to its Parent, which in turn distributed the shares to its members. This issuance of non-voting common stock was accounted for as stock split and no proceeds were received by the Company. The Company also issued to its board of directors, a consultant, and an employee an additional 51,250 total shares in 2022 and recorded $168 thousand of expense for the issuance of these shares, which was based upon a third-party valuation of the shares at the time of issuance.
In December 2022, the Company amended its Certificate of Incorporation to authorize 2,500,000 shares of Series “B” Preferred Stock and issued 381,658 shares in exchange for $2,150 thousand. The Series “B” Preferred Stock has a liquidation preference after Series “A” Preferred Stock and prior to Common Stock and Non-Voting Common Stock. The Series “B” Preferred Stock has voting rights and will automatically convert into Common Stock upon closing of an IPO transaction, as defined in the Company’s Amended and Restated Certificate of Incorporation.
In January 2023, to reflect the ROFN Agreement with its Parent, as further discussed in Note 10, the Company amended its Certificate of Incorporation to a) eliminate upon IPO, Direct Listing, or Sale of the Company the Series “A” Preferred Stock $35,000 thousand liquidation preference, b) make the Series “B” Preferred Stock liquidation preference equal to Series “A” Preferred Stock, and c) to provide that upon IPO, Direct Listing, or Sale of the Company Series “A” Preferred Stock will be canceled for no consideration.
In April 2023, the Company amended its Certificate of Incorporation to authorize 10,000,000 shares of Common Stock, increase the number of authorized Series “B” Preferred Stock shares up to 5,000,000 shares, and to authorize 5,000,000 shares of Series “B-1” Preferred Stock with liquidation preference equal to the Series “A” and “B” Preferred Stock. The Series “B-1” Preferred Stock has voting rights and will automatically convert into Common Stock upon closing of an IPO transaction, as defined in the Company’s Amended and Restated Certificate of Incorporation.
During the first nine months of 2023, the Company raised $4,620 thousand, net of fees, through a Regulation CF offering with 890,310 shares of Series “B” Preferred Stock issued, raised $10,325 thousand in a private placement with 1,680,084 shares of Series “B” Preferred Stock issued, and raised $1,193 thousand in a private placement with 74,922 shares of “B-1” Preferred Stock issued and 8,890 warrants issued subsequent to September 30, 2023.
In October 2023, the Company amended and restated its certificate of incorporation with the State of Delaware to increase to 100,000,000 shares its authorized shares of voting common stock, par value $0.00001 per share, reduce to 30,000,000 shares its authorized shares of non-voting common stock, par value $0.00001 per share, and authorize 2,500 shares of Series C Preferred Stock, par value $0.00001 per share. The Series C Preferred Stock ranks senior to common stock and non-voting common stock and junior to the Series A Preferred Stock, Series B Preferred Stock and Series B-1 Preferred Stock upon liquidation, dissolution, winding-up or otherwise. The Series C Preferred Stock shall have no voting rights prior to an IPO, and 13,000 votes per share upon closing of an IPO. The Series C Preferred Stock is not entitled to dividends, has a liquidation preference of $18.00 per share, subject to adjustment, may be converted 1:1 at any time at the option of the holder into common stock, and upon closing of an IPO will if transferred automatically convert 1:1 into common stock.
| 7. | Share Subscription Agreement |
On November 12, 2021, the Company entered into a Share Purchase Agreement with certain investors for the sale of up to $100,000 thousand of common stock (the “Aggregate Limit”). This agreement is contingent upon the Company achieving a public listing of its common stock. Major terms of the agreement include a commitment fee of 2% of the Aggregate Limit, which is due no later than one year after public listing even if no drawdowns are taken, and five-year warrants issued to the investors at the time of public listing to purchase common stock shares equal to 4% of the total equity interests of the Company at the lesser of a) the price per share at the time of the public listing or b) the quotient of $700,000 thousand divided by the total number of equity interests (fully diluted common shares). The Company may request a drawdown, or sale of common stock shares to the investors, over the five-year term of this agreement following the public listing unless terminated earlier. The amount of the drawdowns requested is limited by the trading volumes of the Company’s common stock shares over the 30-day period preceding the drawdown, and the price per share is equal to 90% of the average price per share over that same period. A 1% fee must be paid to the investors if the Company is sold in a private sale transaction rather than completing a public listing of its shares.
| F-26 |
| 8. | Income Taxes |
A reconciliation of the income tax benefit computed using the federal statutory income tax rate to the Company’s effective income tax rate is as follows:
| Nine
Months Ended September 30, 2023 |
Nine
Months Ended September 30, 2022 |
|||||||
| Federal statutory rate | (21.0 | )% | (21.0 | )% | ||||
| Permanent items | — | — | ||||||
| True up prior year net operating loss deferred tax asset | — | (8.7 | ) | |||||
| Other changes | 0.2 | (1.2 | ) | |||||
| Change in valuation allowance | 20.8 | 30.9 | ||||||
| Total | 0.0 | % | 0.0 | % | ||||
The components of the Company’s net deferred tax assets are as follows:
| (in thousands of dollars) | September 30, 2023 | December 31, 2022 | ||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 1,862 | $ | 896 | ||||
| Lease liability | 378 | 435 | ||||||
| Capitalized research and development | 614 | 299 | ||||||
| Derivative liability | — | 31 | ||||||
| Accrued liabilities | 124 | 81 | ||||||
| Stock compensation | 99 | 17 | ||||||
| Deferred tax assets | 3,077 | 1,759 | ||||||
| Deferred tax liabilities: | ||||||||
| Lease right-of-use asset | (400 | ) | (462 | ) | ||||
| Unamortized debt discount | — | (31 | ) | |||||
| Deferred tax liabilities | (400 | ) | (493 | ) | ||||
| Less: valuation allowance | (2,677 | ) | (1,266 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
The Company was initially formed as an LLC and was converted to a Delaware corporation in December 2021. As a result of generating net operating losses during the years ended December 31, 2022 and 2021, the Company had no income tax expense for years ended December 31, 2022 and 2021. As of September 30, 2023, the Company had U.S. federal net operating loss (“NOL”) carryforwards of $8,867 thousand. The federal NOL carries forward indefinitely and may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest. This could limit the amount of tax attributes that can be utilized annually to offset future taxable income or tax liabilities. Subsequent ownership changes may further affect the limitation in future years.
Effective for tax years beginning after December 31, 2021, taxpayers are required to capitalize any expenses incurred that are considered incidental to research and experimentation (“R&E”) activities under Internal Revenue Code (“IRC”) Section 174. While taxpayers historically had the option of deducting these expenses under IRC Section 174, the December 2017 Tax Cuts and Jobs Act mandates capitalization and amortization of R&E expenses for tax years beginning after December 31, 2021. Expenses incurred in connection with R&E activities in the United States must be amortized over a 5-year period if incurred, and R&E expenses incurred outside the United States must be amortized over a 15-year period. R&E activities are broader in scope than qualified research activities considered under IRC Section 41 (relating to the research tax credit). For the year ended December 31, 2022, the Company performed an analysis based on available guidance and determined that it will continue to be in a loss position even after the required capitalization and amortization of its R&E expenses. The Company will continue to monitor this issue for future developments, but it does not expect R&E capitalization and amortization to require it to pay cash taxes now or in the near future. The Company has included the impact of this provision, which resulted in a deferred tax asset of approximately $614 thousand as of September 30, 2023.
| F-27 |
Management has evaluated the positive and negative evidence bearing upon the realizability of the Company’s net deferred tax assets and has determined that it is more likely than not that the Company will not recognize the benefits of the net deferred tax assets. As a result, the Company has recorded a full valuation allowance at September 30, 2023, and December 31, 2022. The Company will continue to assess the realizability of its deferred tax assets going forward and will adjust the valuation allowance as needed.
As of September 30, 2023, and December 31, 2022, the Company had no uncertain tax positions. The Company recognizes both interest and penalties associated with unrecognized tax benefits as a component of income tax expense. The Company has not recorded any interest or penalties for unrecognized tax benefits since its inception.
| 9. | Leases, Commitments and Contingencies |
The Company has elected in accordance with ASC 842-20-25-2 an accounting policy to not record short-term leases, defined as those with terms of 12 months or less, on the Condensed Balance Sheets. Rent expense recorded under leases, for financial statement purposes, is recognized on a straight-line basis over the lease term based on the most recent contractual terms available.
As of December 31, 2021, the Company had entered into two short-term lease agreements for lab and office space. The Company opted on March 30, 2022, to extend the lease term for one of its leases for lab and office space, with a commencement date for the lease extension on May 1, 2022. The extended lease term was 126 months and was accounted for as an operating lease under the ASC 842 guidance for lease accounting. A right-of-use lease asset and tenant improvement allowance receivable with a combined total of $2,799 thousand and a lease liability of $2,799 thousand were recorded at the time of the extension. This lease was terminated as of July 31, 2022, and the remaining balances of the right-of-use asset, tenant improvement allowance receivable, and lease liability were written off.
The Company expanded the scope and extended for six months the term for the remaining lease for temporary lab and office space on July 1, 2022, then further expanded the scope on August 1, 2022, and October 7, 2022, which increased the monthly license fee to $15 thousand per month. In January 2023, the Company extended this lease for an additional six months and it expired at the end of June 2023. This lease for temporary lab and office space was accounted for as a short-term lease.
In October 2022, the Company entered into a lease agreement for office space with a term of 62 months, which expires on November 30, 2027. This lease will be accounted for as an operating lease under the ASC 842 guidance for lease accounting. A right-of-use lease asset and lease liability of $2,293 thousand each were recorded at inception of the lease term using a discount rate of 7.5%.
In June 2023, the Company entered into a new lease for temporary lab and office space for its research operations. This lease has a term of 12 months and monthly rent of $6 thousand and will be accounted for as a short-term lease. This lease commenced in August 2023. In September 2023, the Company entered into an amendment of this lease to additional space, and the monthly rent increased to $7 thousand.
Rent expense for the nine months ended September 30, 2023 and 2022, was $504 thousand and $198 thousand, respectively. As of September 30, 2023, noncancelable lease payments under operating leases were $2,104 thousand and noncancelable lease payments under short-term leases were $65 thousand.
As of September 30, 2023, future minimum payments during the remaining period and the next five years are as follows (in thousands):
| (in thousands of dollars) | ||||
| 2023 | $ | 86 | ||
| 2024 | 477 | |||
| 2025 | 488 | |||
| 2026 | 544 | |||
| 2027 | 509 | |||
| Thereafter | — | |||
| Total lease payments | 2,104 | |||
| Less: imputed interest | (304 | ) | ||
| Total lease liabilities | 1,800 | |||
| Less: current lease liabilities | (353 | ) | ||
| Total non-current lease liabilities | $ | 1,447 | ||
| F-28 |
| 10. | Related Party Transactions |
As of December 31, 2021, the Company had an outstanding related party Parent company payable of $225 thousand. The debt was held by FibroGenesis, and was due April 1, 2022, with a six-month extension option available at the discretion of FibroBiologics. The Company repaid in April 2022 the remaining Parent company payable of $225 thousand. In July 2022, the Company loaned $300 thousand to the Parent on a one-year note bearing no interest. In October 2022, the Company loaned $60 thousand to the Parent on a one-year note bearing no interest and this note was repaid before December 31, 2022. In April 2023, FibroGenesis repaid in full the $300 thousand Parent company receivable.
As described in Note 6, the Company acquired from FibroGenesis certain in-process research and development and patent assets through Patent Assignment and Intellectual Property Cross-License Agreements. The Patent Assignment Agreement transferred the right, title and interest in and to certain patents from FibroGenesis to the Company for further development. The Intellectual Property Cross-License Agreement grants to the Company exclusive rights to patents owned by FibroGenesis in a limited field of use, which includes the diagnosis, treatment, prevention and palliation of a) spinal diseases, disorders, or conditions, b) cancer, c) orthopedics diseases, disorders or conditions, and d) multiple sclerosis.
In January 2023, the Company entered into an Agreement Regarding Right of First Negotiation (“ROFN Agreement”) with its Parent, FibroGenesis. In exchange for FibroGenesis’ consent to amend the Certificate of Incorporation to a) eliminate upon IPO, Direct Listing, or Sale of the Company the Series A Preferred Stock $35,000 thousand liquidation preference, b) make the Series B Preferred Stock liquidation preference equal to Series “A” Preferred Stock, and c) to provide that upon IPO, Direct Listing, or Sale of the Company Series A Preferred Stock will be canceled for no consideration, FibroBiologics agreed to pay to FibroGenesis 15% of the gross proceeds from any equity investments in FibroBiologics prior to an IPO, Direct Listing or Sale of the Company. In addition, FibroBiologics will receive a five-year right of first negotiation if FibroGenesis decides to license externally any of its technology. In January 2023, the Company amended its Certificate of Incorporation to reflect these changes, recorded a derivative liability of $2,573 thousand for the expected future payments to FibroGenesis, and paid $323 thousand to FibroGenesis for 15% of the gross proceeds from equity issued by the Company in December 2022. Based on its relationship to Series A Preferred Stock as described above, the derivative liability was recorded first against the net Parent Investment and then to Additional paid-in capital after the net Parent Investment was eliminated. During the first nine months of 2023, the Company has raised gross proceeds of $16,418 thousand from equity issuances and has paid $2,322 thousand of these proceeds to FibroGenesis. Amounts paid in excess of the derivative liability are recorded as other losses in the Condensed Statements of Operations. There was no derivative liability and a payable of $141 thousand to FibroGenesis as of September 30, 2023.
| 11. | Share-based Compensation |
The Company adopted on August 10, 2022, and the stockholders approved on August 18, 2022, the 2022 Stock Plan (the “Plan”). The Plan provides for the grant of incentive stock options, nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, and other stock awards. The Plan, through the grant of stock awards, is intended to help the Company secure and retain the services of eligible award recipients, provide incentives for such persons to exert maximum efforts for the success of the Company and provide a means by which the eligible recipients may benefit from increases in value of the common stock. In September 2022, the Company issued a total of 101,250 options with a strike price of $3.28 per share to employees, directors, and scientific advisory board members under this Plan. In February 2023, the Company issued an additional total of 3,689,750 options with a strike price of $2.28 per share to employees and directors under this Plan. In August 2023, a total of 2,500 options with a strike price of $3.28 per share were forfeited. Generally, awards granted by the Company vest over four years and have an exercise price equal to the estimated fair value of the common stock as determined by the board of directors with consideration given to contemporaneous valuations of the Company’s common stock prepared by an independent third-party valuation firm.
As of September 30, 2023, and December 31, 2022, there were 8,711,500 and 12,398,750 shares, respectively, available for future issuance under the Plan.
| F-29 |
Stock-based compensation expense is recognized in the Condensed Statements of Operations as follows:
|
For the Nine Months Ended September 30, |
||||||||
| (in thousands of dollars) | 2023 | 2022 | ||||||
| Research and development | $ | 196 | $ | 100 | ||||
| General, administrative and other | 1,137 | 133 | ||||||
| Total stock-based compensation expense | $ | 1,333 | $ | 233 | ||||
Stock-based compensation expense for the nine months ended September 30, 2022, includes $168 thousand of expense for non-voting common stock issued to the Board of Directors and consultants.
Unrecognized stock-based compensation costs related to unvested awards and the weighted-average period over which the costs are expected to be recognized as of September 30, 2023, are as follows:
| Stock Options | ||||
| Unrecognized stock-based compensation expense (in thousands) | $ | 5,470 | ||
| Expected weighted-average period compensation costs to be recognized (years) | 3.2 |
A summary of the Company’s stock option activity is as follows:
| Stock Options | Weighted-Average Exercise Price per Share | Weighted-Average Remaining Contractual Life (years) | Aggregate Intrinsic Value (in thousands) | |||||||||||||
| Outstanding as of December 31, 2022 | 101,250 | $ | 3.28 | 9.7 | — | |||||||||||
| Granted | 3,689,750 | $ | 2.28 | 10.0 | — | |||||||||||
| Exercised | — | $ | — | — | — | |||||||||||
| Forfeited/Canceled | 2,500 | $ | 3.28 |
9.0 | — | |||||||||||
| Outstanding as of September 30, 2023 | 3,788,500 | $ | 2.36 | 9.4 | — | |||||||||||
| Exercisable as of September 30, 2023 | 56,298 | $ | 3.28 | 9.0 | — | |||||||||||
The fair value of stock options granted to employees, directors, and consultants was estimated on the date of grant using the Black-Scholes option pricing model using the following assumptions:
| Assumptions: |
Nine Months Ended September 30, 2023 |
|||
| Risk-free interest rate | 3.9 | % | ||
| Expected volatility | 90 | % | ||
| Expected term (years) | 7.0 | |||
| Expected dividend | 0 | % | ||
During the nine months ended September 30, 2023, the weighted-average grant date fair value of the options granted was $1.80 per share.
| 12. | Subsequent Events |
The Company has evaluated subsequent events through November 27, 2023, the date the Unaudited Condensed Financial Statements were available to be issued, and has determined that there were no other events, other than what is disclosed in Notes 2 and 6 and below, which occurred requiring disclosure in or adjustments to the Unaudited Condensed Financial Statements.
In November 2023, the Company issued a total of 14,859 additional shares and 10,321 warrants to investors who subscribed to purchase shares of Series B-1 Preferred Stock.
| F-30 |
, 2023
Through and including , 2023 (the 25th day after the listing date of our common stock), all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution |
The following table sets forth the costs and expenses payable by us in connection with this registration statement and the listing of our common stock. All amounts shown are estimates except for the SEC registration fee and the Nasdaq listing fee.
| Amount | ||||
| SEC registration fee | $ | 10,969 | ||
| Nasdaq listing fee | 270,000 | |||
| Legal fees and expenses | 310,000 | |||
| Accounting fees and expenses | 222,000 | |||
Advisory fee |
200,000 | |||
| Printing and engraving expenses | 6,000 | |||
| Transfer agent fees and expenses | 12,850 | |||
| Miscellaneous expenses | 5,000 | |||
| Total | $ | 1,036,819 | ||
| Item 14. | Indemnification of Directors and Officers |
We are incorporated under the laws of the State of Delaware. Section 145 of the DGCL provides that a Delaware corporation may indemnify any person who was or is, or is threatened to be made, a party to any threatened, pending, or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person is or was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with such action, suit or proceeding, if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe the person’s conduct was unlawful.
Section 145 of the DGCL also provides that Delaware corporation may indemnify any person who was or is, or is threatened to be made, a party to any threatened, pending, or completed action or suit by or in the right of the corporation by reason of the fact that such person is or was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification of any claim, issue or matter is permitted without judicial approval if such person is adjudged to be liable to the corporation.
Under the DGCL, where a present or former officer or director is successful on the merits or otherwise in the defense of any action referred to above, or in defense of any claim, issue or matter therein, the corporation must indemnify such present or former officer or director against the expenses (including attorney’s fees) which such present or former officer or director actually and reasonably incurred in connection with such action (or claim, issue or matter therein).
| II-1 |
Section 102(b)(7) of the DGCL permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duties as a director, except for liability for any:
| ● | breach of a director’s duty of loyalty to the corporation or its stockholders; | |
| ● | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| ● | unlawful payment of dividends or unlawful stock purchase or redemption; or | |
| ● | transaction from which the director derived an improper personal benefit. |
Our amended and restated certificate of incorporation which will become effective in connection with the effectiveness of the registration statement of which this prospectus forms a part, will contain a provision that precludes any director of ours from being personally liable to us or our stockholders for monetary damages for breach of fiduciary duty as a director, except for the aforementioned liabilities which we are not permitted to eliminate or limit under Section 107(b)(7) of the DGCL.
In addition, our amended and restated certificate of incorporation and bylaws, in each case, which will become effective in connection with the effectiveness of the registration statement of which this prospectus forms a part, will require us to indemnify, and advance expenses to, to the fullest extent permitted by law, any person who was or is, or is threatened to be made, a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative by reason of the fact that the person is or was our director, officer, employee or agent, or is or was serving at our request as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise.
Our amended and restated bylaws which will become effective in connection with the effectiveness of the registration statement of which this prospectus forms a part, will further authorize us to purchase and maintain insurance on behalf of any person who is or was our director, officer, employee or agent, or is or was serving at our request as a director, officer, employee or agent of another corporation, partnership, joint venture, trust, enterprise or nonprofit entity against any liability asserted against such person and incurred by such person in any such capacity, or arising out of such person’s status as such, whether or not we would have the power to indemnify such person against such liability under the provisions of the DGCL.
We plan to purchase an insurance policy covering our officers and directors with respect to certain liabilities, including liabilities arising under the Securities Act, or otherwise. In addition, in connection with the effectiveness of the registration statement of which this prospectus forms a part, we intend to enter into separate indemnification agreements with each of our directors and executive officers.
| Item 15. | Recent Sales of Unregistered Securities |
The following sets forth information regarding all unregistered securities we have issued since our inception.
Series A Preferred Stock
In connection with our formation, on April 8, 2021, we issued the equivalent of 8,750,000 shares of our Series A Preferred Stock to FibroGenesis in return for rights to certain intellectual property through the Patent Assignment Agreement and the Intellectual Property Cross-License Agreement. See “Business—Intellectual Property” for additional details. No underwriters were involved in the sale of the securities. The sale of the securities were deemed to be exempt from registration pursuant to Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, as transactions by an issuer not involving a public offering.
Non-Voting Common Stock
In January 2022, we issued an aggregate of the equivalent of 37,500 shares of our non-voting common stock for no cash consideration to five of our independent directors, the equivalent of 7,500 shares each, for their service on our board of directors. The issuance of the securities was deemed to be exempt from registration pursuant to Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, as transactions by an issuer not involving a public offering.
In March 2022, we issued the equivalent of 12,500 shares and 1,250 shares, respectively, of our non-voting common stock for no cash consideration to Dr. An and Dr. Khoja for services provided. The issuance of the securities was deemed to be exempt from registration pursuant to Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, as transactions by an issuer not involving a public offering.
In August 2022, we issued the equivalent of 28,179,592 of our nonvoting common stock to our parent company, FibroGenesis. The sale of the securities was deemed to be exempt from registration pursuant to Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, as transactions by an issuer not involving a public offering.
Series B Preferred Stock
In December 2022, we issued an aggregate of the equivalent of 381,658 shares of Series B Preferred Stock to investors in a private placement, at a price the equivalent of $6.76 with respect to the equivalent of 318,049 shares, with the remaining equivalent of 63,609 shares being bonus shares. The sales of the securities were deemed to be exempt from registration pursuant to Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, as transactions by an issuer not involving a public offering.
From February 2023 through April 2023, we issued an aggregate of the equivalent of 890,310 shares of our Series B Preferred Stock to investors in a Regulation Crowdfunding offering, at a price the equivalent of $6.76 as to the equivalent of 724,937 shares, with the remaining equivalent of 143,225 shares and equivalent of 22,148 shares being bonus and commission shares, respectively. The sales of the foregoing securities were issued pursuant to the exemption provided by Section 4(a)(6) of the Securities Act.
In March and April 2023, we issued an aggregate of the equivalent of 1,680,084 shares of our Series B Preferred Stock to investors in private placements, at a price the equivalent of $6.76 as to the equivalent of 1,527,349 shares, with the remaining equivalent of 152,735 shares being bonus shares. The sales of the securities were deemed to be exempt from registration pursuant to Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, as transactions by an issuer not involving a public offering.
Series B-1 Preferred Stock
From April 2023 through September 2023, we issued an aggregate of the equivalent of 74,922 shares of our Series B-1 Preferred Stock to investors in a private placement, at prices ranging from the equivalent of $18.00 to $20.00 per share as to the equivalent of 64,070 shares, with the remaining equivalent of 10,852 shares being bonus shares. In connection with a portion of such private placement of our Series B-1 Preferred Stock, we also agreed to issue warrants, exercisable for a period of three years from their issuance date, to purchase an aggregate of the equivalent of an aggregate of 8,890 shares of our common stock at an exercise price of the equivalent of $20.00 per share. The sales of the securities were deemed to be exempt from registration pursuant to Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, as transactions by an issuer not involving a public offering. In November 2023, the Company issued a total of 14,859 additional shares of Series B-1 Preferred Stock and 1,431 additional warrants to purchase shares of common stock to investors who subscribed to purchase shares of Series B-1 Preferred Stock at a price per share that exceeded the reference price per share expected in the Direct Listing.
| Item 16. | Exhibits and Financial Statement Schedules |
Exhibits
See the Exhibit Index immediately preceding the signature page hereto for a list of exhibits filed as part of this registration statement, which Exhibit Index is incorporated herein by reference.
Financial Statement Schedules
All financial statement schedules are omitted because the information called for is not required or is shown either in the financial statements or in the accompanying notes.
| II-2 |
| Item 17. | Undertakings |
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement.
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
Provided, however, that paragraphs (a)(1)(i), (ii), and (iii) of this section do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the SEC by the registrant pursuant to Section 13 or Section 15(d) of the Exchange Act, that are incorporated by reference in the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(5) That, for the purpose of determining liability of the registrant under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
| II-3 |
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
(b) Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
(c) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(d) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
| II-4 |
EXHIBIT INDEX
| II-5 |
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the city of Houston, State of Texas, on December 1, 2023.
| FibroBiologics, Inc. | ||
| By: | /s/ Pete O’Heeron | |
| Pete O’Heeron | ||
| Chief Executive Officer | ||
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||
|
/s/ Pete O’Heeron |
Chairperson and Chief Executive Officer | December 1, 2023 | ||
| Pete O’Heeron | (Principal Executive Officer) | |||
|
/s/ Mark Andersen |
Chief Financial Officer |
December 1, 2023 | ||
| Mark Andersen | (Principal Financial Officer and Principal Accounting Officer) | |||
|
* |
Director | December 1, 2023 | ||
| Robert Hoffman | ||||
|
* |
Director | December 1, 2023 |
||
| Victoria Niklas, M.D. | ||||
|
* |
Director | December 1, 2023 | ||
| Richard Cilento | ||||
|
* |
Director | December 1, 2023 | ||
| Stacy Coen | ||||
| * | Director | December 1, 2023 | ||
| Matthew Link |
| *By: | /s/ Mark Andersen | |
| Name: | Mark Andersen | |
| Title: | Attorney-in-fact |
| II-6 |